

Yasuhiro Arimura
@ArmrYshr
Followers
904
Following
969
Media
64
Statuses
551
Elucidating the structural basis of biological events on chromosomes | chromatin, Xenopus egg extract, MagIC-cryo-EM | Assistant Professor at @HutchBasicSci
Seattle, WA
Joined September 2018
Don't wanna be here?
Send us removal request.
Explore trending content on Musk Viewer
#Mステ
• 394828 Tweets
#Mステ
• 394828 Tweets
billie
• 285835 Tweets
CHASING THE SUN
• 131835 Tweets
Scottie
• 97954 Tweets
Valhalla
• 42825 Tweets
#のらりクラり呑もうの会
• 35886 Tweets
Louisville
• 31391 Tweets
ひーくん
• 28890 Tweets
ジュラシックワールド
• 16989 Tweets
トップバッター
• 15114 Tweets
しーちゃん
• 14549 Tweets
Mリーグ
• 12843 Tweets
Go City Go
• 12792 Tweets
ベビモン
• 11446 Tweets
NEW hair
• 10988 Tweets
佐々木朗希
• 10591 Tweets
西洸人くん
• 10402 Tweets
#大倉忠義オンライン飲み会
• 10120 Tweets
BACK TO THE BEGINNING
• 10006 Tweets




















Pinned Tweet

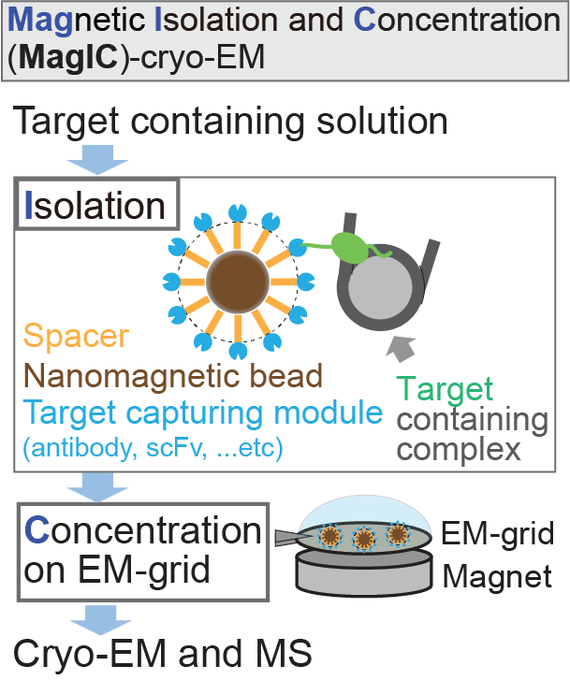
Magnetic Isolation and Concentration (MagIC)-cryo-EM is a new method to enable direct structural analysis of targets captured on nanomagnetic beads, which requires only 0.0005 mg/mL of targets in 100 µL crude solution to make 10 cryo-EM grids. I will explain the magic behind it!
9
118
430
Hey chromatin folks! I am incredibly excited to share my first preprint paper and first paper from the Funabiki lab with you! "Near-atomic resolution nucleosome structures and their variations in interphase and metaphase chromosomes"

10
73
378
Excited to share my final PostDoc work! Introducing MagIC-cryo-EM🪄 & DuSTER🧹, these methods reduce cryo-EM sample requirements (0.0005 mg/mL), enabling ALL biologists to explore the structural basis of diverse biological events!
BioRxiv:

5
69
248
Please repost!
Arimura Lab is hiring! We are seeking the next generation of MagICians (technicians and a postdoc) to elucidate the structural basis of events on chromosomes🧙♀️
No prior structural biology experience is required!!
Link

Magnetic Isolation and Concentration (MagIC)-cryo-EM is a new method to enable direct structural analysis of targets captured on nanomagnetic beads, which requires only 0.0005 mg/mL of targets in 100 µL crude solution to make 10 cryo-EM grids. I will explain the magic behind it!
9
118
430
0
105
165
I am extremely excited to start my lab at Fred Hutch next year! Perfect environment to study chromatin! My lab aims to elucidate the structural basis of biological events on chromatin through the structural analyses of "native" chromatin complexes!

2023 was such an amazing year for the division, including the launch of the
@FredHutch
Postbac Program, the hiring of three of the brightest minds in their fields (
@C_P_Lapointe
,
@SRsrivatsan
,
@ArmrYshr
), and so much more. Read our 2023 Annual Report:
0
8
18
5
16
163
Plasmids for MagIC-cryo-EM are now available from Addgene! 🧙♀️
Magnetic Isolation and Concentration (MagIC)-cryo-EM is a new method to enable direct structural analysis of targets captured on nanomagnetic beads, which requires only 0.0005 mg/mL of targets in 100 µL crude solution to make 10 cryo-EM grids. I will explain the magic behind it!
9
118
430
2
23
128
My second paper (preprint) from Funabiki lab!! Actually, it's not chromatin work. We reported the cryo-EM structure of alpha-2-Macroglobulin (A2M) co-fractionated with the nucleosome. This work revealed the mechanism of how A2M captures proteases.
1/n
6
22
125
I am so excited that I can finally share the peer-reviewed version of my native nucleosome cryo-EM paper!

Online Now: Structural features of nucleosomes in interphase and metaphase chromosomes
0
28
83
9
22
113
I am thrilled to begin my lab at my dream institute! 😆😆Recruitment ads will be posted in a couple of weeks. Stay tuned!
We are incredibly excited to welcome our newest faculty member,
@ArmrYshr
, on his first day in Basic Sciences
@FredHutch
! The Arimura Lab will study how chromatin functions, how it’s regulated, and what goes wrong in diseases like cancer. Learn more:
2
3
19
0
13
98
I am so excited to present in the Fragile nucleosome seminar!

Happy Friday everyone! Next week's seminar is going to be fantastic! We'll get to hear from two amazing structural biologists
@ArmrYshr
and
@lapassmore
!! **Recommended to attend live** Register here:

0
22
61
2
13
60
Check out the other new paper published today from
@KurumizakaLab
!
I've contributed to the first part of the work. Probably this one is my favorite Fig 1 among my works.
Everything started from a total failure experiment...
3
12
58
My preprint was selected as a "Promising Preprints"😂 Thank you, folks!

Near-atomic resolution nucleosome structures and their variations in interphase and metaphase chromosomes
0
0
3
3
1
56
Stunning work by my Ph.D. mentor, Kurumizaka-sensei! He started RecA (bacterial homolog of Rad51) research during his Ph.D. work at the Shibata lab. Then, he started nucleosome research at the Wolffe lab. Now, these two finally unite in the cryo-EM structure! Congratulations!🎉

Our new paper about the structural mechanism by which RAD51 assembles at double-stranded DNA break sites on chromatin has been published in Nature!
#CryoEM
#Nucleosome
#RAD51
#DNArepair
11
121
514
0
7
56
I studied about AlphaFold last night for the journal club today. To me (zero-background of protein structure prediction), the AlphaFold paper was nearly impossible to understand. But these materials helped me a lot to capture the principle of it.
1
13
50
Hey, chromatin enthusiasts in New York! I will present my data on the NYC chromatin club next Tuesday. See you at the meeting! (I am retweeting this to push me to prepare my presentation😅)
January 12, 2021 5-6PM EST
Speakers
Bobbie Pelham-Webb (Weill-Cornell)
“The molecular resetting of pluripotent stem cell identity upon mitotic exit”
Yasuhiro Arimura (Rockefeller University)
“Nucleosome structural variations in interphase and metaphase chromosomes”
1
0
13
2
10
49
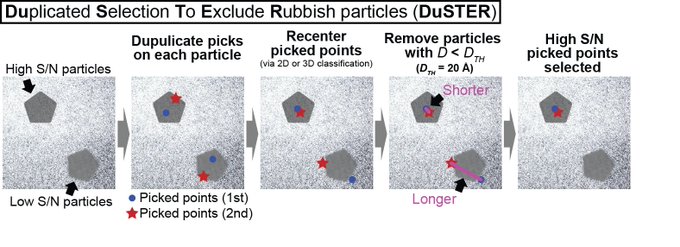
Duplicated Selection To Exclude Rubbish particles (DuSTER) is a method for facilitating the structural analysis of small biomolecules. DuSTER uses the reproducibility of the particle centering during 2D classification to exclude the low S/N.
1
15
44
I'm thrilled to be part of the MCB program and help the next generation of talent thrive!

Join us in welcoming our newest faculty members, Dr. Yasuhiro Arimura (
@ArmrYshr
) from
@HutchBasicSci
and Dr. Devasena Ponnalagu (
@DPonnalaguLab
) from
@UWPharmacology
!


0
1
8
0
5
45
Mission complete😎
My second paper (preprint) from Funabiki lab!! Actually, it's not chromatin work. We reported the cryo-EM structure of alpha-2-Macroglobulin (A2M) co-fractionated with the nucleosome. This work revealed the mechanism of how A2M captures proteases.
1/n
6
22
125
5
3
41
We determined the near-atomic resolution structure of nucleosome in interphase and metaphase chromosome by combining the Xenopus egg extract system I have learned at the
@HironoriFunabi1
and the nucleosome structural biology I have learned at the
@Kurimizakalab
.
1
5
31
Check out the exciting paper from my fantastic colleague!! My latest preprint showed that the H1 tightly bound on the nucleosome dyad in the M phase. He has revealed the novel H1 function in the M phase: controlling the number of condensins and topo II on chromosomes!

Happy to share our preprint, by which we propose the novel molecular function of the linker histone H1 in regulation of the mitotic chromosome shape and organization. Here I introduce a historical background for a broad audience. 1/
13
40
149
0
7
30
I am on YouTube now😳
Just in time for the weekend! If you'd like to watch back Yasuhiro Arimura's talk on cryo-EM structures of nucleosomes in interphase and metaphase, you can find it on our YouTube channel! Thank you for sharing your work in our series,
@ArmrYshr
!
0
5
22
0
5
23
Cool!! ScFv for the acidic patch can somehow stabilize H1 on dyad (does it reduce NCP structural variation??). Also, I've never expected that H3 N-terminal goes this orientation in the chromatosome.

With the aid of an scFv fragment. We have reconstructed cryo-EM maps of four chromatosomes at 2.8-3.1 Å. all H1 isoforms show similar on-dyad binding mode, though the local interactions between H1 isoforms and nucleosome are divergent. 1/7
1
7
36
2
3
23
Arimura Lab is also participating in the Fred Hutch Postbaccalaureate Scholar Program! The scholar develop a foundation for a research career through chromatin biology research in our lab🧙🧙♂️🧙♀️

The Fred Hutch Postbaccalaureate Scholar Program is accepting applications! This program is for recent college grads who would benefit from research experience and training prior to applying to
#biomedical
PhD programs. Apply by Monday, April 1:

0
17
13
0
6
17
H4-H4 four-helix bundle!! Super cool! Congratulations Nozawa-san, Kurumizaka-Sensei!!

最新論文をbioRxivにて公開しました!
Cryo-electron microscopy structure of the H3-H4 octasome without histones H2A and H2B
2
9
48
1
2
21
To improve my Twitter environment, I've made a bot to review English words/idioms I came across in PostDoc life in the US
@StudyingYasu
Feel free to follow it if you are also an English learner🤓
0
2
16
I previously found that the CENP-A nucleosome forms outward H4 tail confirmation and this confirmation somehow facilitates H4K20me1 modification by SET8(AKA PR-SET7)
This work explained why. H4 tail orientation in CENP-A nucleosome is ready to bind SET8!!
Our SET8-nucleosome cryo-EM structures are finally published in Life Science Alliance!
The structures reveal how SET8 recognizes and binds the nucleosome substrate to induce efficient mono-methylation of H4K20.
2
14
56
1
3
14
Super cool!!!😍😍
0
2
14
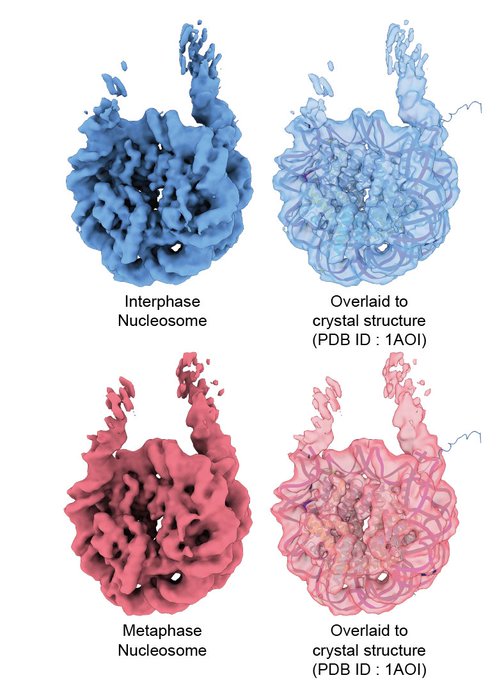
Unlike alpha2-macroglobulin, the average structures of "in chromosome" nucleosomes were perfectly matched with the crystal structure of the nucleosome (Fig. 2, 3). (Probably a good news for nucleosome folks! )
2
0
12
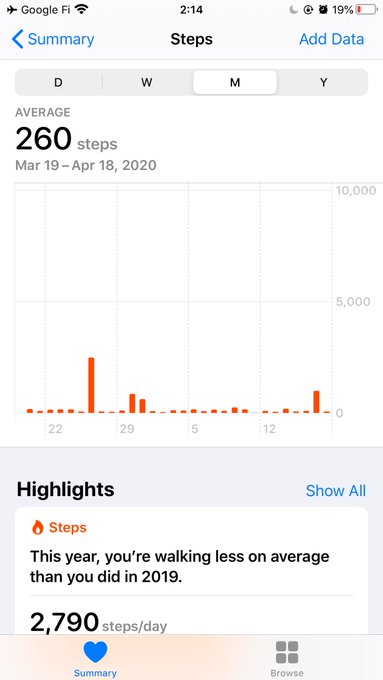
My average steps this month! 260 steps/day! Can I be more quarantined?!
0
0
11
Check out my colleague's Tweetorial (Xtorial?) about her amazing finding: the identification of the new hemimethylated CpG binding protein!

Very excited to announce our pre-print! Together with the labs of
@NishiyamaAtsuya
and
@KyoheiArita
, we’ve discovered that the protein CDCA7 (a long-time favourite in the Funabiki lab) exhibits a striking ability to sense hemi-methylated DNA. 1/7
4
9
41
0
0
11
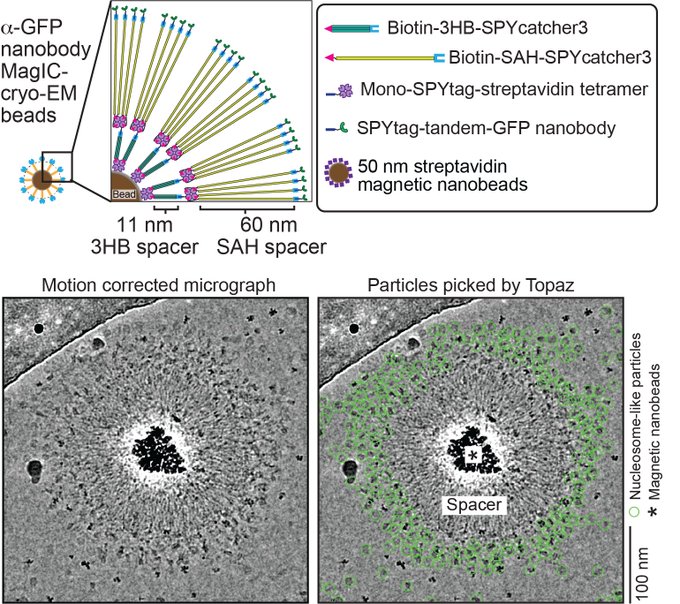
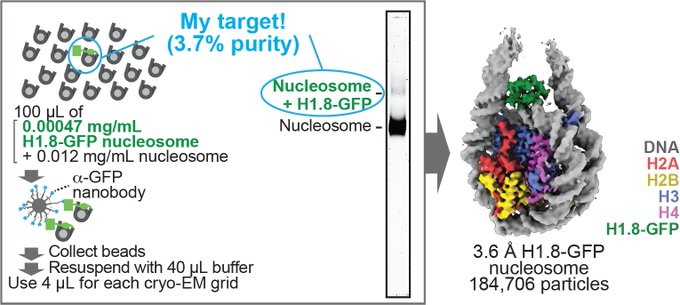
After years-long optimization, we could solve this issue by attaching the spacer proteins to beads! Importantly, the target-capturing module can be replaced with any proteins. In this work, we used GFP nanobody.
1
1
11
The latest collaboration work with my undergrad lab, where I graduated 13 years ago☺️
Drs. Ohnuki and Takano did an MD simulation of peptide synthesis by RimK based on our published X-ray structure. Our new X-ray structure supported their model.
0
0
10
Virus nucleosome week!🤯🤯

Indeed, some viruses encode histone doublets!
#viralnucleosomes
are fragile, and have extended linker DNA. Check out the structures. AND they are specifically recruited to the Marseillevirus factory and virus, shown by our collaborators
@chantal_abergel
.
.
6
32
159
0
0
10
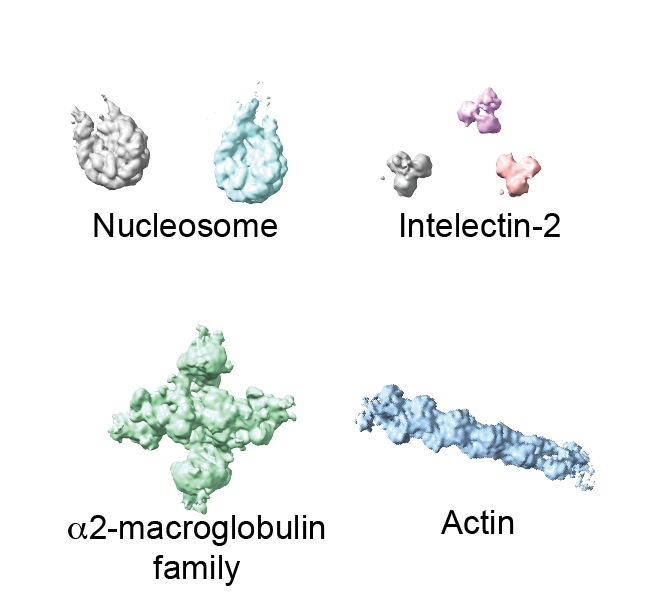
With tremendous help from Seth Darst lab and
@RubyFroom
in his lab, we determined the structures of the five most abundant complexes (nucleosome, chromatosome, alpha2-macroglobulin, intelectin-2, actin filament) at once in an unbiased way (Fig. 2).
1
0
10
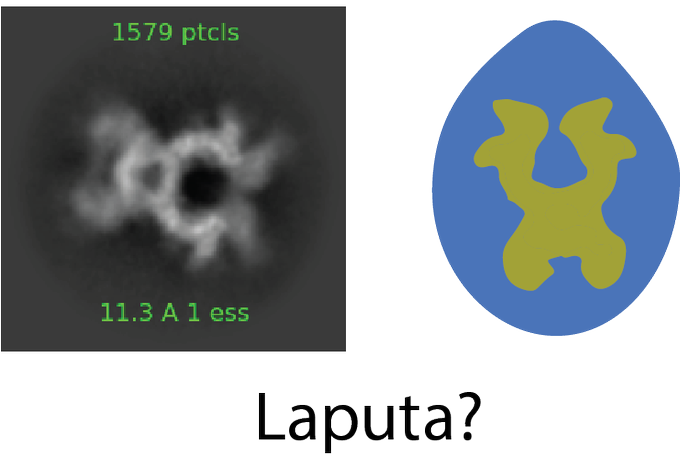
Although I personally name this protein "Laputa" because the 2D class is reminiscent of the crystal necklace in the Ghibli movie Laputa, I had no idea what the real name of this protein was.
6/n
1
1
10
Please read my preprint!

Near-atomic resolution nucleosome structures and their variations in interphase and metaphase chromosomes
0
5
23
1
2
9
I used this 3DVA in my latest preprint. The 3DVA (& cryoSPARC of course) is amazing🤯 Thank you for developing this awesome method!!

Interested in continuous heterogeneity+flexibility in
#cryoEM
? Check out our updated
#cryoSPARC
3D Variability Analysis paper on
@biorxiv
!
It’s great to see 3DVA being used in the wild to resolve beautiful proteins in motion 😀
1
14
57
1
3
9
By selecting the particles that are reproducibly re-centered by 2D and 3D classification, we could dramatically improve the 2D maps and successfully determine the structure of the interphase-specific H1-containing complex!
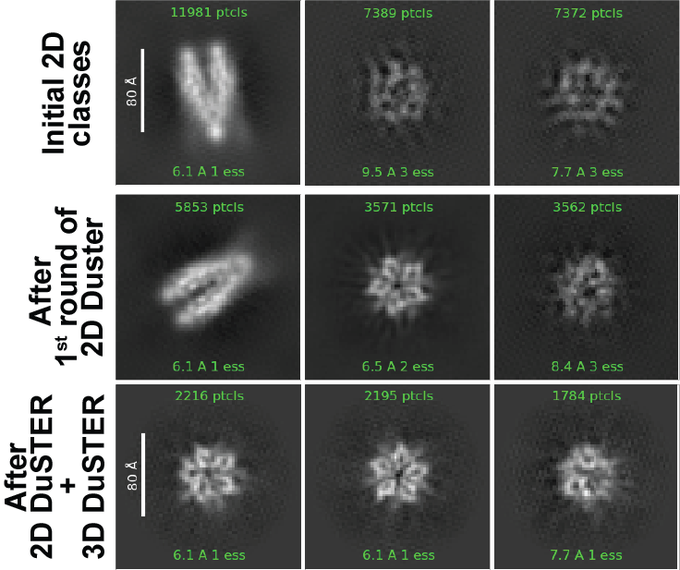
2
2
8
Congratulations, Daniel🥳
The cryo-ET structure of the native kinetochore is more flexible and larger than anticipated!
(Also, MagIC-cryo-EM contributed in an unanticipated way: makes proper thickness ice)

The bulk of my grad work is finally out in the world! Big thanks to everyone in
@SueBiggins
lab and collaborators including
@HHMIJanelia
,
@WijeratneS
,
@ArmrYshr
,
@HironoriFunabi1
, and others not on this site. Wouldn't have been possible without great collaborators!
1
11
30
0
0
8
We hope this study become the first step to link the function and structure of the nucleosome in the chromosome.
3
0
8
Up until now, no method has been established to analyze the high-resolution nucleosome structure in chromosomes, and the function of the nucleosome structure and its variant in chromosomes can not be addressed.
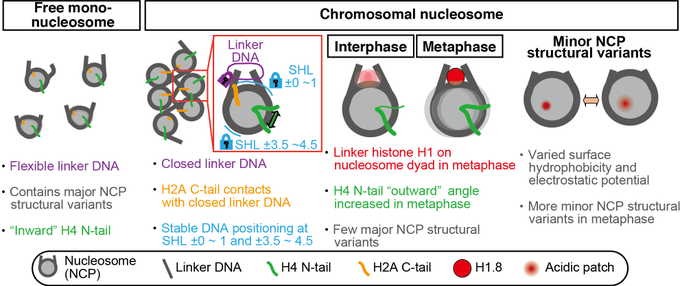
1
0
8
A really cool work from Seth Darst's lab (one of my cryo-EM mentors)!! Congratulations🎊

Early intermediates in bacterial RNA polymerase promoter melting visualized by time-resolved cryo-electron microscopy
#bioRxiv
0
1
5
0
0
7
Christian is an amazingly kind man with a broad range of profound knowledge. I do recommend this position!

Post-doc position available in my lab for a collaborative project together with Pascal Meier
@MeierLabICR
and Jon Pines
@PinesLab
to study innate immune responses to radiation. Apply at or message me for more information.
4
48
68
1
2
8
Using Xenopus egg extract, we made functional interphase and metaphase chromosomes on the Xenopus genome DNA and isolated the crosslinked interphase and metaphase chromosomes with the exact identical procedure. Then determined the nucleosome structures at up to 3.4Å resolution.
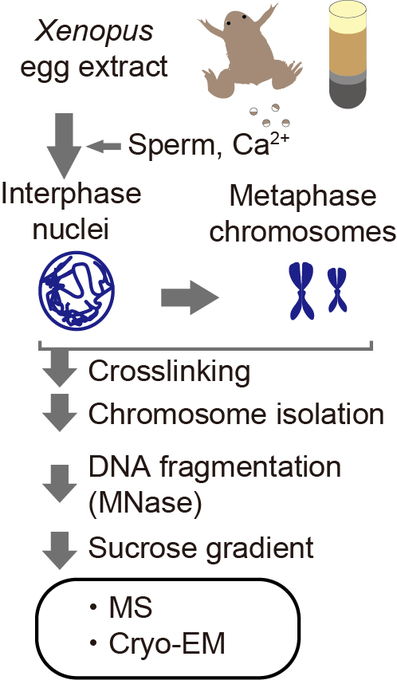
1
0
8
I am currently updating this method to visualize many other chromatin targets for understanding chromatin functionalities by angstrom-level structure. I hope I can share my next work soon! [end of the thread]
1
0
7
Unexpectedly, exclusively for metaphase nucleosome, we found the linker histone H1.8 at the on-dyad position (Fig 4) and determined the H1.8 bound nucleosome structure at 4.4 Å resolution (Fig. 6).
1
0
7
I will send plasmids to addgene today. Please try it in your lab!
1
0
6
Actually, I wasn’t sure if I should publish this. I felt guilty while I was writing this paper. I wanted to use my time for chromatin stuff (and job search). But I felt more guilty about not sharing this with the public. So, I decided to do it as my summer vacation activity.
25/n
1
1
6
I've almost forgotten to explain our finding... We found that nucleosomal H2A.B exchanges with canonical H2A without additional factors! Congratulations Hirano-san!
0
0
6
Here are the links for my Tweetorial!! I hope you enjoy reading them!
MagIC:
Magnetic Isolation and Concentration (MagIC)-cryo-EM is a new method to enable direct structural analysis of targets captured on nanomagnetic beads, which requires only 0.0005 mg/mL of targets in 100 µL crude solution to make 10 cryo-EM grids. I will explain the magic behind it!
9
118
430
1
2
6
One is the "major structural variants," which are characterized by jagged NCP outlines with sliding of the H3-H4 tetramer, as compared to the smoother circular shape of the canonical NCP. Our data suggest that this type of movement is suppressed in the chromosome (Fig. 7).
1
0
6
Thank you for many comments to our preprint!! We modified the title.
Thanks for overwhelmingly positive responses to our recent preprint by
@ArmrYshr
!
>1200 downloads!
In response to a comment by
@RadoDanev
, we uploaded a V2 with a modified title.
Nucleosome structural variations in interphase and metaphase chromosomes
1
1
46
0
0
6
So, we next focused on a small interphase-specific complex that showed up in the sucrose gradient. Here, we realized another big limitation of the cryo-EM: the size. The structural analysis of the molecule <100kDa is very challenging. We devised DuSTER to address this issue!
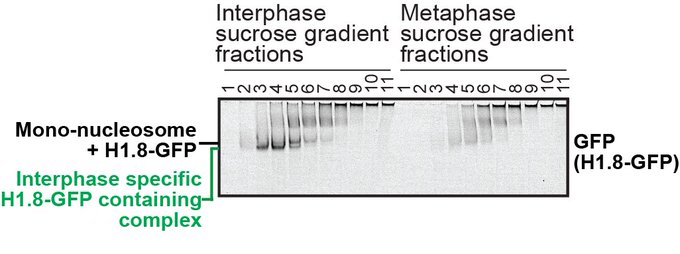
1
0
2
We were inspired by the highly creative work published in 2010. In this study, the authors isolated virus pseudoparticles using nanomagnetic beads and directly applied them to cryo-EM grids to take snapshot images.

1
0
6
Since alpha2-macroglobulin is the extracellular protein, we have never expected that we determine this protein's structure. Interestingly, the structure of "native" alpha2-macroglobulin was dissimilar to the crystal structure of the recombinant protein.
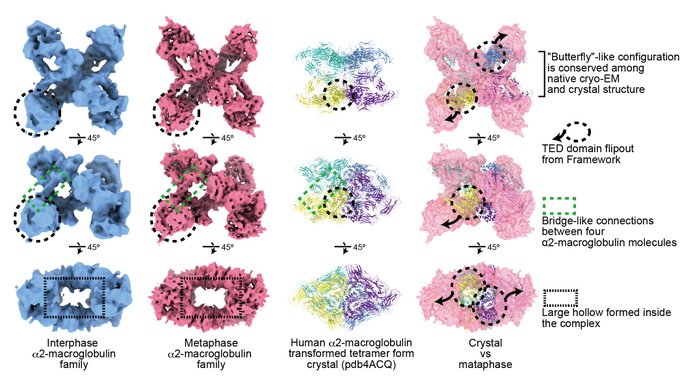
1
0
6
Wow😍

Welcome check out our new manuscript. DISCA: high-throughput cryo-ET structural pattern mining by deep unsupervised clustering.
The work was led by my PhD student Xiangrui, in collaboration with
@yiweichang
and Mahamid teams
@Liang_Xue_
.
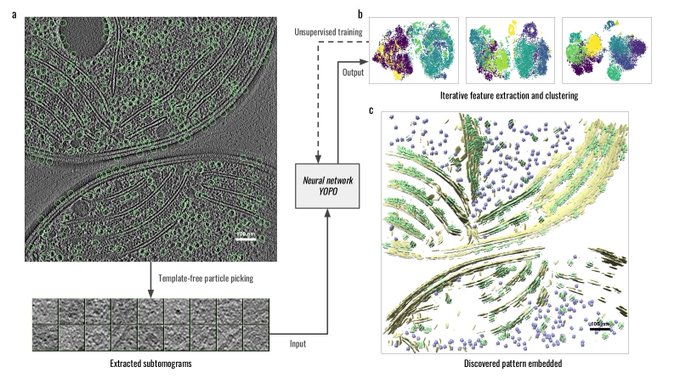

1
26
93
0
0
6
I thank my super supportive mentor
@HironoriFunabi1
, who kindly learned about A2M from scratch with me with turning himself into a Dracula.
.
26/n
I feel like I'm a Dracula as I feel my energy slowly coming back while I chew my postdoc's exciting manuscript w/o being exposed to the direct sunlight for last 4 days...
1
0
10
1
1
5
To address this, we should isolate specific complexes for cryo-EM. Traditional methods, like ChIP, can achieve isolation, but cryo-EM's demanding 0.1~5 mg/mL sample requirement posed challenges for my sample. Our goal is to reduce it to ChIP-seq levels, widely used in our field.
1
0
4
I hope my dear chromatin people enjoyed this tweetorial, and real alpha-2-macroglobulin scientists notice this work😅
end/n
1
0
5
🤯

Please see our Cryo-EM structure of nucleosome-like particles formed by Marseillevirus histone fusions and DNA online in
@NatureSMB
. Fantastic work by
@Marco_Va1
and
@Xagbomson
! Special thanks to our collaborators! Here is a link:
15
69
273
0
0
5
@HironoriFunabi1
Thank you so much for your support! The five years I spent in the Funabiki lab were amazing. Combining your expertise with mine, we developed unique approaches. You also connected me with so many scientists. Thank you so much!
1
0
5
'The H2B-E76K mutant fundamentally alters chromatin accessibility' The authors reported the effect of H2B-E76K mutant in cells that I really wanted to find and I couldn't in my previous work! Great!
A Mutation in Histone H2B Represents a New Class of Oncogenic Driver.
0
4
3
0
1
5
Molecular replacement of
crystallization contaminants👀
AlphaFold Protein Structure Database for Sequence-Independent Molecular Replacement
#bioRxiv
0
18
81
0
0
5
Amazing!!🤯

We’re excited to share cryo-TomoSim (aka CTS), our
#cryoET
modeling and simulation software, in addition to our new preprint documenting its use in training regressive denoising and semantic segmentation U-Nets.
7
72
334
1
0
5
(if you have a friend working on alpha2-macroglobulin, please let them know that the "native" alpha2-macroglobulin structure (Fig S3G) is available now. )
1
0
5
This paper is SO exciting for a nucleosome structure freak!!
Many nucleosome X-ray crystallography studies showed that DNA length in the nucleosomes was not uniform (144-147bp).
3
2
5
Then, to understand the structural variety of the "in chromosome" nucleosome, we used the 3DVA of cryoSPARC (Fig. 4).
1
0
5
Using cryo-EM maps and mass spec, we identified this molecule as NPM2 and found a novel conformation of NPM2 (open form) that might be important for the H1 association!
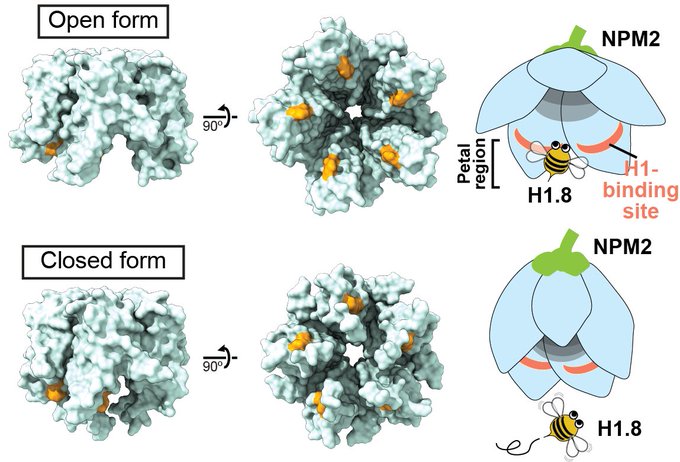
1
0
3
To avoid human bias, we did NOT choose nucleosome-like 2D classes and used all blob-picked particles for ab initio and heterogeneous reconstruction of cryoSPARC. Thanks for the great capability cryoSPARC, several different cryo-EM structures at once.
4/n
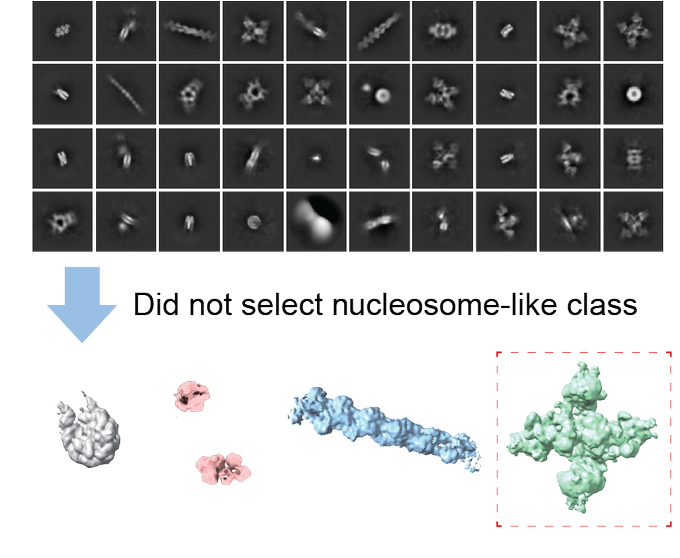
2
0
5
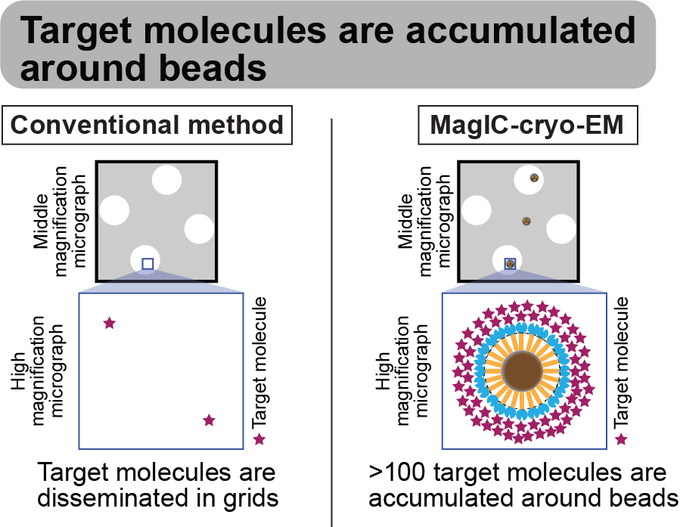
Trick 4: Targets are highly concentrated around the beads, ensuring that each micrograph contains more than 100 usable particles for 3D structure determination.
1
0
5
Trick 3: The magnetic beads are easily identified in middle-magnification montage maps, enabling the selection of areas where targets exist prior to high-magnification data collection.

1
0
5
This is so cool!!!

Next, we use in situ sequencing to read out the libraries via fluorescence microscopy.
This is a single nucleus: each spot is an amplified DNA fragment, the colors indicate the 4 DNA bases, and each frame represents a round of sequencing (3/8)
1
6
47
0
0
5
To avoid human bias, we did NOT choose nucleosome-like 2D classes and used all blob-picked particles for ab initio and heterogeneous reconstruction of cryoSPARC. Thanks for the great capability cryoSPARC, several different cryo-EM structures at once.
4/n
2
0
5
1
0
4
Compared to the free mono nucleosome, both interphase and metaphase nucleosomes had more closed linker DNA and stable nucleosome core particle structure (Fig. 4). Our results suggest that there is an active mechanism to close the linker DNA of nucleosomes in chromosomes (Fig. 5)
1
0
4
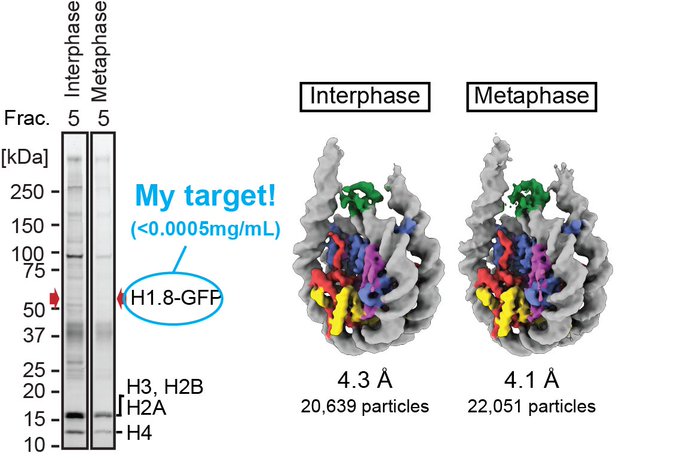
Using MagIC-cryo-EM, we could determine the structures of H1.8-GFP nucleosomes in interphase and metaphase. Notably, the concentration of my target is 0.0002~0.0005 mg/mL, and the sample is very heterogeneous.
1
0
4
Using these beads, we tested with in vitro reconstituted nucleosomes if we could target scarce macromolecules in heterogeneous samples. As a result, we could determine the H1.8-GFP nucleosome structure from a heterogeneous solution (4% purity) with 0.0005 mg/mL of targets🪄
1
0
4
Also, this work was only possible because I was trained by the nucleosome enthusiast, Kurumizaka-sensei
@KurumizakaLab
during my Ph.D. training.

1
0
4
My paper was selected as the "Editors' Highlights" paper of Nature Communications!!
0
0
4
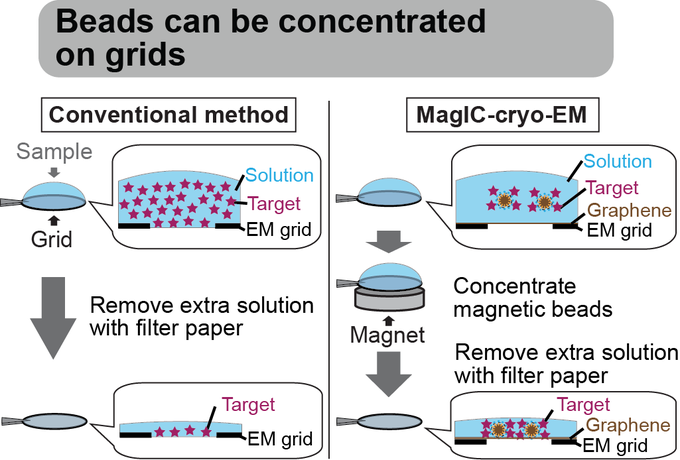
Trick 2: Sample loss during the grid-freezing process is reduced by magnet-based enrichment of the targets on cryo-EM grids.
2
0
4
We tested if this approach can be used for the single particle analysis to determine 3D structures. While we got a promising result with 50 nm avidin beads, it revealed the big problem of this approach: a strong background noise generated around the magnetic beads.
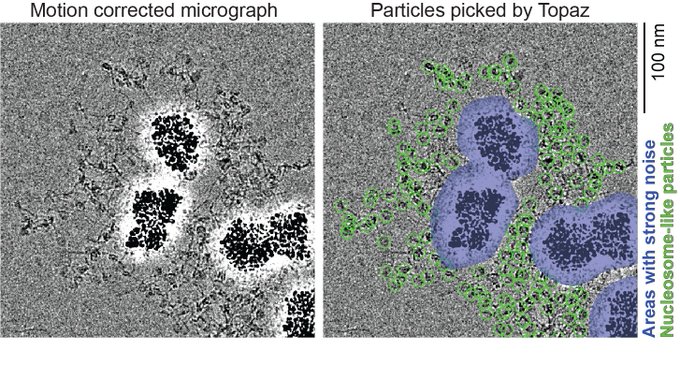
1
0
4
or the youtube video at Fragile Nucleosome Seminar Series!
Just in time for the weekend! If you'd like to watch back Yasuhiro Arimura's talk on cryo-EM structures of nucleosomes in interphase and metaphase, you can find it on our YouTube channel! Thank you for sharing your work in our series,
@ArmrYshr
!
0
5
22
1
1
3
In minor NCP structural variants, H4 N-terminal tail orientation, surface hydrophobicity, and electrostatic potential are changed. These changes may affect the inter-nucleosome interaction and protein binding on nucleosomes and contribute to chromatin compaction in the M phase.
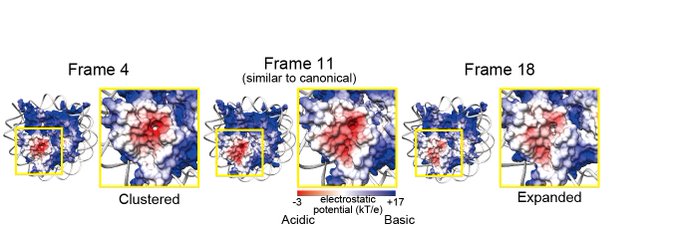
1
0
3
@SaKanYoo1
通りすがりです。プラスミド同士が複数くっつくことは、昔ながらの方法で検出できないだけで、実はよく
あるみたいです。 ただ、例に挙げられた二つの制限酵素は、巨大な回文配列になってしまうので、大腸菌の中で生き残らないのではないでしょうか。

I think
@plasmidsaurus
is just trolling me 💀"Propagate a plasmid in MG1655, what's the worst that can happen?"
5
3
40
1
1
3
Thank you for reading such a long Tweetorial. I hope this low sample requirement democratizes structural analysis for ALL biologists to explore the structural basis of diverse biological events!
1
0
3
DuSTER:
Duplicated Selection To Exclude Rubbish particles (DuSTER) is a method for facilitating the structural analysis of small biomolecules. DuSTER uses the reproducibility of the particle centering during 2D classification to exclude the low S/N.
1
15
44
2
0
3
We showed the mechanism that enables the H4K20me1 accumulation on CENP-A nucleosome by X-ray crystallography, biochemistry (collaborate with Kimura Lab
@Cell_Tokyo_Tech
), and Quantitative ChIP-Seq (collaborate with Fukagawa Lab
@tatsuofukagawa1
).
1
0
3
@RubyFroom
@HironoriFunabi1
Thank you so much for your advice and coaching on data processing!! The analyses in the paper were only possible because of your help!!
0
0
3
@LugerLab
@UCSFChimeraX
@biorxivpreprint
Wait.. is it a split nucleosome?? I can't wait to read the paper
0
0
3
The second question is how the cleavage of the A2M bait region by protease cause the transformation of A2M. This mechanism is conserved among A2M homologs that do not form a tetramer.
21/n
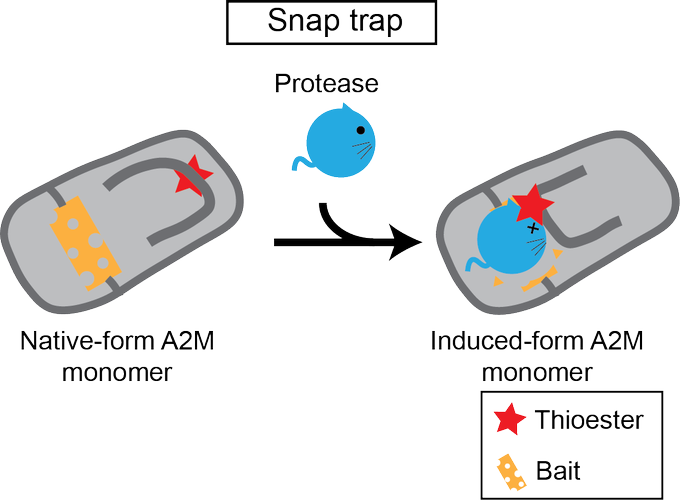
2
0
3
@LugerLab
@DShechter
Thank you for your interest! Here is a link for signing up for the NYC chromatin club mailing list to get the zoom link (Please hit "subscribe" on the top left). I assume the club is open for worldwide chromatin people since the club tweet this public 🤔
NYC Chromatin Club - Jan 12, 2021 at 5PM -
Subscribe for details and a zoom link!
0
0
6
3
0
3
Congratulations!! So cool work!! Nucleosome structure folks might love Fig 6😁
1
0
3
In our previous work, we determined the structures of the nucleosomes and H1-bound nucleosomes formed in chromosomes. However, >300 other chromatin-associated proteins detected by MS remained unsolved.
Hey chromatin folks! I am incredibly excited to share my first preprint paper and first paper from the Funabiki lab with you! "Near-atomic resolution nucleosome structures and their variations in interphase and metaphase chromosomes"
10
73
378
1
0
3