

amelie_schreiber
@amelie_iska
Followers
1,020
Following
343
Media
27
Statuses
568
I ❤️ proteins! Researching protein language models, equivariant transformers, LoRA, QLoRA, DDPMs, flow matching, etc. intersex=awesome😎✡️🏳️🌈🏳️⚧️💻🧬❤️🇮🇱
California
Joined May 2023
Don't wanna be here?
Send us removal request.
Explore trending content on Musk Viewer
Ayuso
• 184593 Tweets
No Riley
• 142264 Tweets
Sean
• 94056 Tweets
#NEDFRA
• 83451 Tweets
Jacob Juma
• 82697 Tweets
Austria
• 78694 Tweets
Francia
• 65112 Tweets
#LondonTSTheErastour
• 60223 Tweets
Olise
• 54646 Tweets
Bayern
• 49058 Tweets
Griezmann
• 39453 Tweets
Kanté
• 37588 Tweets
Rabiot
• 37243 Tweets
オランダ
• 30546 Tweets
فرنسا
• 30288 Tweets
Thuram
• 29193 Tweets
Holanda
• 26050 Tweets
Dembele
• 22724 Tweets
Clancy
• 22528 Tweets
Deschamps
• 21079 Tweets
The Dutch
• 18313 Tweets
DIAN
• 15037 Tweets
Barcola
• 14815 Tweets
THE BLACK DOG
• 13954 Tweets
HITS DIFFERENT
• 13886 Tweets
#PBSFRA
• 13291 Tweets
Depay
• 13145 Tweets
كانتي
• 13067 Tweets
Xavi Simons
• 10298 Tweets
Maroon
• 10282 Tweets




















Top 10 ❤️ tools rn, in no particular order:
1. ProteinDT
2. MoleculesSTM
3. RFDiffusion-AA
4. RosettaFold-AA
5. LigandMPNN
6. Distributional Graphormer (DiG)
7. DNA-Diffusion
8. OAReactDiff
9. RFDiffusion (original)
10. EvoDiff
❤️ Evo
❤️ Flow matching
❤️ Boltzmann generators
2
27
196
Protein binding a small molecule designed with RFDiffusion-AA yesterday. I'm such a huge fangirl for these all-atom models. Baker Lab is awesome!

2
17
170
These two together make a really good pair:
From this you get conformational ensembles and binding affinity for protein-protein, protein-small molecule, and protein-nucleic acid affinities, reducing the need for expensive MD sims.

0
24
138
Found out yesterday some of my
@huggingface
blogs inspired some undergrads to start studying AI applied to proteins and someone applied to and received an internship based on their interest in replicating and extending some of them. 😎 Feeling very inspired and grateful now. ❤️
4
8
130
Hope this helps 😊

2
33
132
In case it is helpful:

1
13
94

Just thought I would share this new Hugging Face community blog post I wrote as a follow up post to the ESMBind post. It explains how to build an ensemble of Low Rank Adaptations (LoRAs) after you have finetuned multiple ESMBind LoRA models:

1
13
57
This just happened.

1
6
56
RNA sequence design analogous to ProteinMPNN, but for RNA:

1
6
55
An interesting and novel approach to applying transformers to graph structured data. This never got the attention it deserved and is likely an approach lost to time. It maybe “old” but it’s worth investigating further, especially for biochem/molecules:

3
8
55
Damn, another E(3)-equivariant model that should have been SE(3)-equivariant. Molecules have chirality! Still exciting that it works for small molecules AND proteins:

0
7
55
Has anyone else tried grafting two proteins together by first placing the proteins into AlphaFold-Multimer, then linking the proteins together with something like RFDiffusion motif scaffolding (treating the two proteins as though they are in the same chain)?
3
5
51
Equivariant Spatio-Temporal Attentive Graph Networks to Simulate Physical Dynamics: A Replacement for MD? TBD. More comments to come.
OpenReview:
GitHub:

4
9
46
BindGPT sounds pretty cool! No code though 😒 probably because they’re on to something with this one, especially when considering the performance and the inference cost drop together. High throughput is really needed for this problem.

3
2
49

1
12
42
Recently wrote a new blog post on intrinsic dimension of protein language model embeddings and curriculum learning:

1
7
41
Working on a new method to cluster protein-protein complexes so I can finetune ESM-2 on them for predicting PPIs and for generating binders 😊. Also may try to finetune EvoDiff this way for generating binders. I ❤️ proteins so much.
2
1
40
Here’s a new method for sampling the equilibrium Boltzmann distribution for proteins using GFlowNets:
If you aren’t familiar with GFlowNets, head over to
@edwardjhu
’s twitter and watch his video. I’ll also post a link to a related lecture soon.

3
4
40
Just cooked up a new tokenization method for protein language models and large language models. I can't wait to share :)
1
1
40
Not specifically for proteins or other molecules, but this is a nice intro to flow matching. Thanks for the video
@ykilcher
any chance you’d ever do something on this applied to proteins?

0
8
38

0
9
34

3
5
35

0
10
31
This looks pretty cool! Also helpful for cutting down on expensive MD simulations. Can't believe I'm just now noticing this work.

1
6
29
Whenever an open source version of
#AlphaFold
3 is being created, be sure to try swapping out the diffusion module for a flow matching module. It’ll probably turn out better that way 😉
2
4
29
ESM-AA huh? Would it be better to use random order autoregressive decoding (similar to LigandMPNN for example) instead of MLM? It seems like a harder objective to train on, but you could end up with a better performing model.
3
3
28
New updates, now we have BioT5+
I'm excited to try this model out.

0
5
21
Let’s go!
“CRISPR-GPT leverages the reasoning ability of LLMs to facilitate the process of selecting CRISPR systems, designing guide RNAs, recommending cellular delivery methods, drafting protocols, and designing validation experiments to confirm editing outcomes.”

2
2
18
Shouldn't we be able to do something similar to this with LoRA?
LoRA and SVD are conceptually very similar. If so, that would likely explain the results in this paper where LoRA turns out to be better than full finetuning Thoughts?

0
1
18
Apparently you can in fact do flow matching on discrete data, for those interested in diffusion applied to discrete data like language and NLP, this is a good reference for how to do it with the more general flow matching models:

Combining discrete and continuous data is an important capability for generative models. To address this for protein design, we introduce Multiflow, a generative model for structure and sequence generation.
Preprint:
Code:
1/8
2
91
445
0
1
17
Okay, hear me out…stratify an NLP dataset (or any other modality really) by using a “homology search” using a BERT style model, similar to this paper, but for non-protein data:
Could help determine the amount of generalization, no?

2
0
15
Another E(3)-equivariant model that should be SE(3)-equivariant. E(3) doesn’t preserve chirality of molecules.
GitHub:


0
1
14

0
1
14
Love child from Distributional Graphormer (
#DiG
) and
#alphafold3
when? C’mon
@Microsoft
and
@GoogleDeepMind
. If
@OpenAI
and
@Apple
can team up to deliver
#Her
we can also have a new model that does dynamics for complexes of biomolecules. You’re almost there 🔥🔥🔥you got this.
3
2
13
Are Kolmogorov-Arnold Networks (KAN) enough to address some problems in biochemistry that suffer from data scarcity? Apparently they require much less data to converge, and all they’re really doing is making activation functions trainable using B-splines.
1
2
11
Thank you for inviting me and for the wonderful conversation.

The AI Revolution in Biology is here - it's just not evenly distributed, even among biologists
@amelie_iska
previews biology as an experimental information science, on the latest Cognitive Revolution – out now!
Listen to catch up!
(link in thread)
2
4
22
2
2
11
Interestingly, quantizing state space models like Mamba doesn't seem to work very well, whereas we are now in the era of 1-bit quantization for transformers ~without~ performance degradation; it also isn't clear if Mamba is as expressive as Transformers.

2
2
11
If you have opportunities to work at the intersection of AI and proteins, DM me. I have ideas and I like implementing them :)
2
5
11
RFDiffusion could’ve been used and it’s suggested this would improve outcomes. Why wasn’t it used for this problem? I’m curious. This would establish more use cases for RFDiffusion and similar methods (like flow matching with FoldFlow-2 for example).

Now online: We developed novel oligomers and turned them into FGFR agonists via binder induced receptor clustering.
#denovo_proteins

3
29
109
1
1
11
(1/n) Even if Sora isn't currently capable of accurately generating simulations of small molecules or proteins, open sourcing it or giving select researcher access to it would allow us to add in equivariance or use components of it such as those that maintain temporal coherence.
4
0
10
Okay, serious question. If you can accomplish the same thing with more general proteins, why restrict yourself to antibodies? Also, what are some problems that really truly require antibodies specifically and that can’t be done with more general proteins?
5
0
10
Seems like an interesting method. I find it very interesting that it works better (SOTA?) if you give it conformational ensembles to work with. Could be very interesting to see how conformational sampling, Distributional Graphormer, or AlphaFlow might yield better results.

Having a lot of fun visualising the ligand binding site predictions of
#IFSitePred
with
#PyMol
! A new ligand binding site prediction method that uses
#ESMIF1
learnt representations to predict where ligands bind! Check it out here:
#Q96BI1
0
2
20
2
1
10

0
2
10
@TonyTheLion2500
I highly recommend this reference along with his “smooth manifolds” book: Introduction to Riemannian Manifolds (Graduate Texts in Mathematics)

0
0
10
Curious to know where this goes.

0
3
9
This looks pretty interesting! Text guided diffusion model for molecules.

0
0
9

Key insight from recent events…patent the method not the molecule. Some of these AI methods are going to wreck patents imo. 🤫
That said…
MIT license >> patent
(for humanity…usually).
2
0
8
@SimonDBarnett
Code is linked to in the Nature paper. This AI model actually samples the Boltzmann distribution, giving all the metastable states (low energy conformations) as well as the transition pathways between them. It’s a “generative diffusion model”:
0
1
8
Goal: use partial diffusion and motif scaffolding to engineer a new version of nitrogenase then modify plant genetics to produce this new version so that chemical fertilizer is unnecessary.

0
0
8

To all those just getting into this stuff: You’re entering one of the most interesting and impactful areas at the most exciting time. Don’t give up, even when it feels impossible. Stay close to the open source biochem AI community. They’re a great crowd. Good luck and have fun!
1
0
7
Having solid temporal coherence, or modifying the architecture to be SE(3)-equivariant would allow us to create better versions of things like this:
and we might actually be able to replace MD with AI, speeding up drug discovery and solving major problems

0
0
6
And it has a BSD-3 license! Not bad.



0
2
7
Crowdsourcing suggestion…if you could selectively disrupt or augment a pathway or PPI network, where would you start? Assume you can block any PPI, or augment the PPI network by designing proteins that create intermediary interactions (ex: proteins that bind/link two others)
3
0
6
@alexrives
I have a method for detecting AI generated proteins that I would like to open source at some point if people are interested. It seems to work on proteins generated by most models out right now, although there are a couple models it does not work for, hesitant to say which ones.
2
0
7
Selectively modulating PPI networks by designing high affinity and high specificity binders with RFDiffusion and checking that with AF-Multimer LIS score seems like low hanging fruit to me. What reasons might there be for this not being very actively & heavily worked on?
2
0
6
Computational efficiency in equivariant models is often a concern. This model addresses that and creates fast SE(n)-equivariant models for tasks involving molecules:
0
1
6

0
2
6
Eeep! It's wooorkiiing! So excited! 😊 I'll write a hf blog post on it once it's all done.
0
1
6
@samswoora
You should also check out flow matching models. Flow matching generalizes diffusion (diffusion is a special case of flow matching). They're doing a lot with proteins and flow matching, but there's less buzz about it in vision and language domains.
2
0
5
Definitely the vibe I’m going for 😂

summer student project presentations are incredible

21
550
11K
0
0
5
🤔

1
0
5
@maurice_weiler
@erikverlinde
@wellingmax
Could someone recommend a similar resource for other architectures like equivariant transformers or equivariance in geometric GNN models? Just curious what the go to resources are for people for other architectures.
2
0
4
Now, using persistence landscapes we can cut down clustering time from a full day to less than 30 minutes for 1000 proteins!

0
2
5
@pratyusha_PS
This is awesome. When will the code be available? I would love to try this with a protein language model like ESM-2 and see if it improves performance.
2
0
5
Attempting to raise my signal to noise ratio today by making some quality posts about AI and biochemistry. 😊
0
0
4
🤔 Based on this lecture (see 20:15), I think cancer is a biological “vector bundle” with degenerate or absent transition functions. In other words, local data dominates and the cohesive global structure does not exist. I wonder how far the analogy goes.

0
0
5
@310ai__
It might also be good to look into computing the LIS score based on the PAE output of RoseTTAFold All Atom similar to what was done with AlphaFold-Multimeter here. This is a new approach for protein-small molecule complexes.
0
1
3
Hot take for some, obvious to others: GPUs and LLM oriented ASICs along with AI operating systems will make CPUs mostly obsolete. Anyone out there capable of writing CUDA kernels who can explain why this might be an erroneous prediction?
2
0
4
@HannesStaerk
@chaitjo
@SimMat20
@ADuvalinho
Just found this and it seems to address some of the concerns over computational cost of equivariant architectures:
0
0
3
This would be cool for proteins
I'd love to try and use this for designing protein-protein complexes in sequence space. Too bad the code isn't released.

2
0
4
Pretty neat. How does it compare to a contrastive model like ProteinDT or ProteinCLIP? And could we use it for annotating in order to train a new ProteinDT or ProteinCLIP? Is there an exceptional text-guided diffusion model coming soon for proteins?

Excited to share new work from
@GoogleDeepMind
: “ProtEx: A Retrieval-Augmented Approach for Protein Function Prediction”

3
41
153
1
0
3
@andrewwhite01
You can also learn equivariance. I think equivariance is an overrated mathematical concept tbh. It's fancy and neat from a mathematical perspective, but otherwise I think you could have your network learn it and get just as far if not further.
0
0
4
This is such a cute animation! 😎 Now do it for 3 modalities like ProTrek (text, protein sequence, protein structure)! 🧬

[CLIP] by Hand ✍️
The CLIP (Contrastive Language–Image Pre-training) model, a groundbreaking work by OpenAI, redefines the intersection of computer vision and natural language processing. It is the basis of all the multi-modal foundation models we see today.
How does CLIP work?
8
195
1K
0
1
5
Anyone who understand the current state of the art in pharmacogenetic testing for determining drug efficacy and side effects, please reach out for discussion or to share some papers. I have questions, and *potentially* a few good ideas on how to improve this.
0
0
3
@befcorreia
@karla_mcastro
@_JosephWatson
@jueseph
@UWproteindesign
Curious to know why RFDiffusion motif scaffolding wasn’t tried here instead of or in addition to the RoseTTAFold constrained hallucination.
1
1
4
Anyone have any idea why in silico directed evolution might increase perplexity and intrinsic dimension of a protein? Are more fit proteins generally more complicated?
3
0
4
I think actually training this model could be done on Lambda Labs for around $150K (20 GPU days on 256 A100s) no? There is a difference between training and inference too that should be made clear here. Inference (using the model for predictions) is much cheaper than training.

Because downloadable code would let 100s of scientists put AlphaFold3 through its paces on different kinds of systems -- for benchmarking and for developing new protocols and code that has the ability to access input parameters (e.g. like colabfold does) missing from the server.
0
1
15
1
2
4
Until everyone can code in natural language, we will not have enough “coders”.
0
0
4
@HannesStaerk
Still REALLY want to see this done with AlphaFold-Multimer. Maybe there’s a dynamic model of PAE and LIS that comes out of this that helps determine how strong or transient a PPI is.
1
2
4
@biorxiv_bioinfo
Cool idea, but how was the dataset split into train, test, and validation? Was sequence similarity/homology used to split the protein dataset? If not, this paper's results are unreliable. You have to split your data based on sequence similarity; 30% similarity is pretty standard
0
0
3
@KKapusniak1
@PPotaptchik
@TeoReu
@leoeleoleo1
@AlexanderTong7
@mmbronstein
@bose_joey
@Francesco_dgv
@FrankNoeBerlin
this maybe useful for transition pathways between conformations.
0
0
4
AlphaFlow-Multimer with the appropriate generalization of the LIS score would more or less solve PPI prediction. LIS alone already mostly solves it. Then the only bottleneck for giant detailed PPI networks is compute. This is a big deal. Explain to me why I might be wrong.
0
0
4
I love this. Thank you! Gotta go watch now! p(shittakes | Michael Levin) << p(shittakes | Amelie Schreiber) 😂 Also,
@drmichaellevin
…feel free to DM anytime with project ideas 🤓
1
0
4
@iScienceLuvr
SVD initialization would’ve helped a lot.
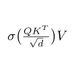
My take on "LoRA Learns Less and Forgets Less"
1) "MLP/All" did not include gate_proj. QKVO, up & down trained but not gate (pg 3 footnote)
2) Why does LoRA perform well on math and not code? lm_head & embed_tokens wasn't trained, so domain shifts not modelled. Also reason why

7
123
574
0
0
3
It would be very interesting and useful to see how this could be used in tandem with the following method for detecting binding sites of conformational ensembles of proteins using ESM-IF1:
0
0
3
@ZymoSuperMan
RFDiffusion works with structures, not sequences. For designing sequences that fold into the backbones that RFDiffusion generates you’ll need something like LigandMPNN which does allow for things like biasing particular residues in various ways to constrain the sequences designed
1
0
3
After that, team up with
@BakerLaboratory
and make the best “RFDiffusion” and “LigandMPNN” anyone’s ever seen, but this time use continuous and discrete flow matching resp. and make it for all the biomolecules. 4 essential “foundation models” and we’ll be all set +/-ε 🎉😎🧬
0
0
3
@lpachter
For academic uses that don’t compete with Isomorphic Labs’ research… that part is subtle, but important. It means if you want to develop a new drug and have any hope of taking it to market, or if you’re not in academia, you’re out of luck. And no reproducing because patents! 😒
0
0
3
Really cool channel. Maybe we’ll get a video on SE(3)-equivariant neural networks one day🤞This would be great for folks trying to understand new SOTA models for proteins and small molecules. I would totally be down to collaborate
@mathemaniacyt
🧬

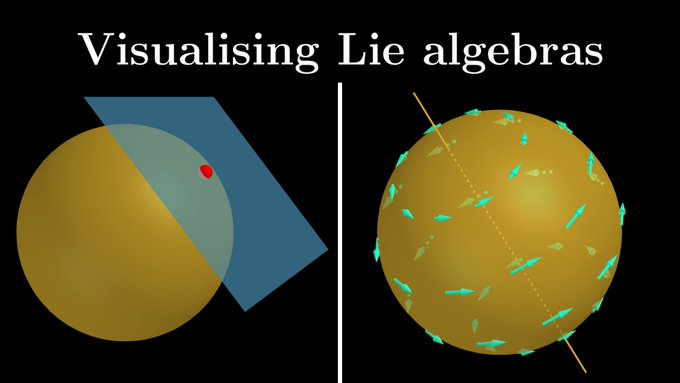
Why do we require Jacobi identity to be satisfied for a Lie bracket? In the process, we also understand intuitively why tr(AB) = tr(BA) without matrix components.
Watch now:
2
103
611
0
0
3