
Bryan Quach @bcquach.bsky.social
@bryancquach
Followers
60
Following
442
Media
2
Statuses
117
Joined October 2015

Here's a new preprint from our group. "Identifying compounds to treat opiate use disorder by leveraging multi-omic data integration and multiple drug repurposing databases." https://t.co/hheSg4cHx6
@carynwillis94 @bryancquach @RTI_Intl

medrxiv.org
Genes influencing opioid use disorder (OUD) biology have been identified via genome-wide association studies (GWAS), gene expression, and network analyses. These discoveries provide opportunities to...
2
5
14

Posting my talk for tomorrow's #ISMBECCB2023 Keynote https://t.co/YFNjS2S183 A Gradual Evolution of Bioinformatics Research: 1994-2052
5
21
86

Prof. @g_parmigiani, Prof. Sur & co-authors researched how to improve cross-study replicability of predictions by providing a theoretical rationale for when to select multi-study ensembling over merging. Read more in their ArXiv paper:

arxiv.org
Cross-study replicability is a powerful model evaluation criterion that emphasizes generalizability of predictions. When training cross-study replicable prediction models, it is critical to decide...
0
3
6

Quantitative assessment of association between noncoding variants and transcription factor binding
0
1
2

Stefano Mangiola @steman_research & I are planning a tutorial on "Tidy Genomic Analysis" Dec 12, 4:30 PM EST Dec 13, 8:30 AM AEDT 60 min + 30 min Q/A - Tidy single-cell and bulk transcriptomics - Tidy enrichment with plyranges and nullranges Sign up: https://t.co/psk4FeLCs6
5
65
175


The MPRA review paper I shared last week was a teaser for this one. We have performed MPRA on ~5,000 schizophrenia finemapped variants and detected ~400 variants with allelic regulatory activity (MPRA-positive variants). https://t.co/MbBk5RoPb4
2
40
167
Is it possible to compare genetic effects across ancestries without biased by environmental and cultural differences? Yes, if the genomic segments representing different ancestries come from the same individuals, i.e., admixed individuals. Brilliant idea! https://t.co/8wlqhkDMfq
medrxiv.org
Individuals of admixed ancestries (e.g., African Americans) inherit a mosaic of ancestry segments (local ancestry) originating from multiple continental ancestral populations. Their genomic diversity...
1
23
91

Excited to announce the release of our new manuscript ( https://t.co/PpofpDEFJS) on the role of mRNA splicing in opioid use disorder. Great to work with @Spencer_Huggett @karlakaun and John McGeary to publish this in Genes @MDPIOpenAccess

mdpi.com
Opiate/opioid use disorder (OUD) is a chronic relapsing brain disorder that has increased in prevalence in the last two decades in the United States.
1
4
16

Thursday, June 9 at 4pm EST: Jeremy M. Simon, PhD (UNC Chapel Hill) leads a #DataScience Zoominar on "Identification of Novel Oncogenic and Neurodevelopmental Programs Using Bulk and Single-Cell Approaches." RSVP: https://t.co/X5rL4qor68
1
3
4

Very grateful to have the opportunity to work with @tianzhang4921 dissecting how genetic variation affects protein phosphorylation. Our great team included @stevemunger, @MTFerris, @GygiLab, and Gary Churchill @jacksonlab. I'll share a few insights from the manuscript.

Excited to share this pre-print on multi-omics analysis identifies drivers of protein phosphorylation in Collaborative Cross mouse strains. My awesome collaborators include @grkeele, Gary Churchill, @stevemunger. @GygiLab
1
9
18

Mathematical expressions can now be displayed in Markdown on GitHub using the $$ and $ delimiters - all with the help of the wonderful @MathJax project.
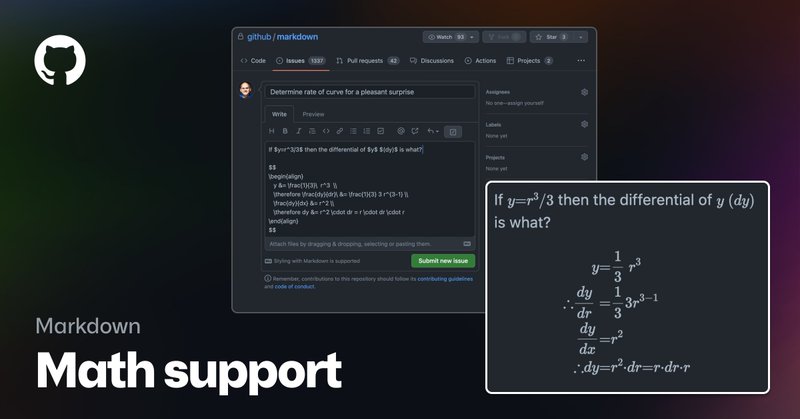
github.blog
We are pleased to announce that math expressions can now be rendered natively in Markdown on GitHub
67
1K
4K

Our paper describing NEAT-seq, a method to simultaneously measure transcription factor protein abundance, ATAC-seq, and RNA-seq in single cells, is now out at Nature Methods! Congrats to Amy and Ben!
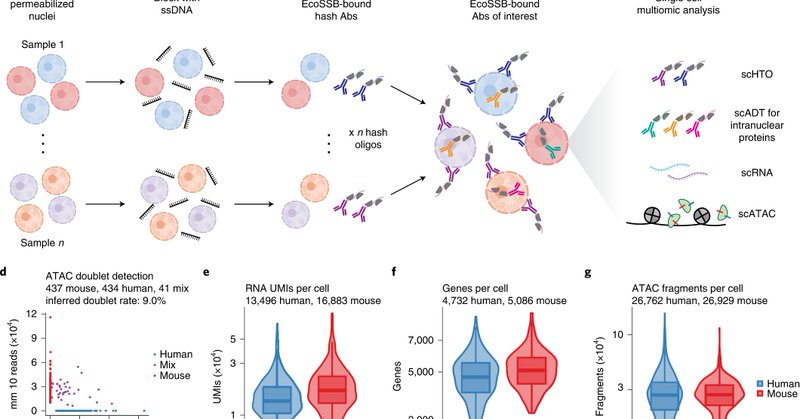
nature.com
Nature Methods - NEAT-seq offers a tri-omics tool for profiling nuclear protein abundance along with ATAC-seq and RNA-seq in single cells.
11
372
2K

The first Corces Lab paper is out today! We've released a step-by-step guide to bulk ATAC-seq @NatureProtocols. This was definitely a labor of love by stellar postdoc Fiorella Grandi.
12
138
584
Got an RNA-seq dataset with 50, 100, 200+ samples? Plug it into a differential expression tool and hope for the best? No! You need to consider QC, EDA, and modeling technical variation, or else risk generating spurious results. A thread on papers, methods, and best practices:
15
420
1K
Please...stop!

I am going to keep bringing up this plea https://t.co/uIKxxSQsY4 as long as people keep using Tophat. This recently published paper used TopHat 1.4.1:
2
4
15

Here we compared 18 functional weighting methods for identifying novel genetic associations and found enhanced discovery for #GWAS ... but such methods are "unlikely to circumvent the need for larger sample sizes in truly underpowered GWAS." @bryancquach et al. @RTI_Intl

Evaluation of methods incorporating biological function and GWAS summary statistics to accelerate discovery https://t.co/CU7OQtCf9V
#bioRxiv
0
3
10

🧠UNC Faculty Recruitment🧬: we are looking for a clinician or physician scientist to be a bridge between Genetics and Psychiatry. We have a fantastic environment of both basic neuro/genetics & clinical expertise. Please RT and distribute widely.
0
7
12

Some extremely interesting results were shown today by @joelhirschhorn on behalf of GIANT consortium (here I use the word GIANT as both noun and an adjective ;)) https://t.co/banp6P8krK

"Computational and functional gene prioritization from a saturated GWAS of adult height in 5 million people" 5 MILLION! Looking forward to this talk by @joelhirschhorn today at #ASHG2021
1
26
65