

Sergei Pond
@sergeilkp
Followers
2K
Following
778
Media
155
Statuses
1K
Dad. Husband. Born/raised Kyiv. Developer of @hyphy_software. Former mathematician, current professor of biology at Temple U. Google scholar: https://t.co/qyzpbkE1qZ
Gladwyne, PA
Joined January 2017
1/ Genome-wide (and other) positive selection screens in genomes may be severely biased by alignment and other errors. we (@averyselberg, @NathanClark111, @ChikinaLab, @nekrut, @stvnwvr, @aglucaci, @timsackton) propose a simple fix.

1
27
89
The 15% indirect cost cap by the NIH is denotating a nuke under the current business model of many US schools, especially medical schools. Major changes incoming. Maybe relaized savings will translate to larger grant budgets/better success rates, but I not holding my breath.
0
3
4
Oops. On the day the new 2024 highly cited researcher list is released, clarivate website crashes. How 1990s.

0
0
1
6/ The new method BUSTED-E, is available in @hyphy_software and will be on soon. Use it by default if you are interested in detecting selection, and also use it to screen alignments for errors before feeding them to other tools.
1
0
7
5/ Instead of detecting ~40-60% of genes under selection, error-corrected methods detect a much more plausible ~5-15%. These genes also make more sense: they are in the more reasonable pathways and functional categories, and less enriched for long genes (~more repeats/error prob).
1
0
6
4/ By extending the BUSTED model in @hyphy_software to "absorb" these error-like parts of the alignment, we kill two birds with one stone: protect positive selection detection from errors and develop an evolutionary-model-based filtering approach.
1
0
3
3/ When this happens, it often looks like a small (<1%, often <0.1%) fraction of the alignment is apparently subject to very strong (dN/dS > 100) selection. More likely, this is not "real" selection, but aberrations, e.g., alignment/homology errors.
1
1
3
2/ Large-scale genome alignment construction uses great filtering tools already, and they do catch many errors. The ones that slip through are "residual" errors. But they are enough to light up sensitive selection detection models in @hyphy_software and other tools.
1
0
3
RT @biorxiv_bioinfo: AUTO-TUNE: SELECTING THE DISTANCE THRESHOLD FOR INFERRING HIV TRANSMISSION CLUSTERS #biorxiv_….
0
3
0
You can now run @hyphy_software entirely in your browser thanks to @emscripten, @observablehq and biowasm (@RobAboukhalil). Simple example in
observablehq.com
`version` Please wait for the version to appear; this will indicate that the .wasm files have loaded `Run on User Data` Code Imports
2
21
62
I like the revised NIH review criteria (. Now please revise grant budget caps to reflect the reality that 250K in 2023 is worth ~50% compared to the 1990s (.
usinflationcalculator.com
Easily calculate how the buying power of the U.S. dollar has changed from 1913 to 2025. Get inflation rates and U.S. inflation news.
0
0
10
This seems highly apropos w.r.t to the posts in my feed regarding the revised Bayes Factors in

science.org
In the Research Article “The molecular epidemiology of multiple zoonotic origins of SARS-CoV-2,” the frequency of simulated topologies matching phylogenetic structures arising from a single introdu...

Bayes factors evaluate priors, cross validations evaluate posteriors.
2
1
8
RT @PhilaUnion: #UthankU giveaway!. RT for a chance to win a signed King Julian Carranza jersey!. #DOOP

0
931
0
3 We offer some implementable recommendations on how this can be done. I think debates like this are increasingly important in the context of large scale genomic pathogen surveillance (see GISAID etc)

0
0
3
2 There is not a particularly good solution. 1. Sequences may be released with no or minimal metadata. This makes them a lot less useful for impactful research. 2. Random subsets of sequences may be released. This does not always preserve privacy AND reduces power.
1
0
2
1 What's the right balance for data sharing vs data privacy? . For HIV, sharing VIRAL genetic data has numerous ethical challenges. HIV-status is PHI. Sequence data can (and have!) been used in criminal transmission cases.
journals.plos.org
Author summary • Human immunodeficiency virus (HIV) drug resistance has implications for antiretroviral treatment strategies and for containing the HIV pandemic because the development of HIV drug...
2
3
10
Great study. Clever genomic forensics. Molnuipravir: playing generic roulette.

📄 📄 Our molnupiravir work is now out after peer-review! We definitively demonstrate that molnupiravir has resulted in viable SARS-CoV-2 viruses with significant numbers of mutations, in some cases with onwards transmission of mutated viruses.
0
7
28
RT @theosanderson: 📄 📄 Our molnupiravir work is now out after peer-review! We definitively demonstrate that molnupiravir has resulted in vi….
nature.com
Nature - A specific class of long phylogenetic branches, distinguished by a high proportion of G-to-A and C-to-T mutations, are almost exclusively found in sequences from 2022, after molnupiravir...
0
607
0
Fascinating work on "generic" antigen design Great to see my former post-doc advisor @sdwfrost continuing to contribute to innovative projects.
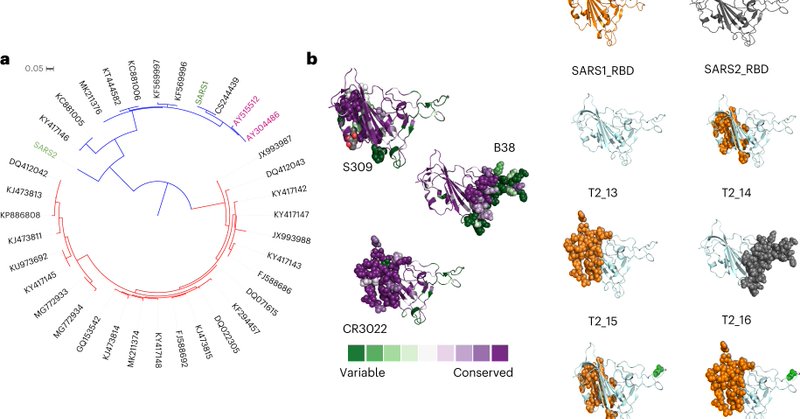
nature.com
Nature Biomedical Engineering - A computationally designed antigen based on the receptor binding domain of the spike protein of sarbecoviruses elicits broad humoral responses against severe acute...
2
6
17