

Timofey Prodanov
@TimofeyProdanov
Followers
62
Following
28
Media
4
Statuses
17
PhD ⋅ PostDoc @UniklinikDUS ⋅ Bioinformatics, Structural Variations, Segmental Duplications.
Düsseldorf, Germany
Joined October 2020
We finally uploaded a preprint on Locityper—targeted genotyper for complex genes, on which @tobiasmarschal and I worked for almost two years!.

biorxiv.org
The human genome contains numerous structurally-variable polymorphic loci, including several hundred disease-associated genes, almost inaccessible for accurate variant calling. Here we present...
2
5
21
RT @MishaKolmogorov: So excited to have this preprint out! Severus is a new tool for somatic SV calling in cancer using long reads. It supp….

medrxiv.org
Most current studies rely on short-read sequencing to detect somatic structural variation (SV) in cancer genomes. Long-read sequencing offers the advantage of better mappability and long-range...
0
29
0
And thank you to our co-authors Elizabeth Plender, Guiscard Seebohm, Sven Meuth and @EichlerLab.
0
0
1
Locityper carefully manages read alignments, insert sizes and read depth profiles to identify the two most likely locus haplotypes (gene alleles) for a given short or long-read WGS dataset.

1
0
0
Parascopy is available on github ( and can be installed with conda (conda install -c bioconda parascopy). Thank you for reading! Please check the paper for more details [7/7].
github.com
Copy number estimation and variant calling for duplicated genes using WGS. - tprodanov/parascopy
0
0
0
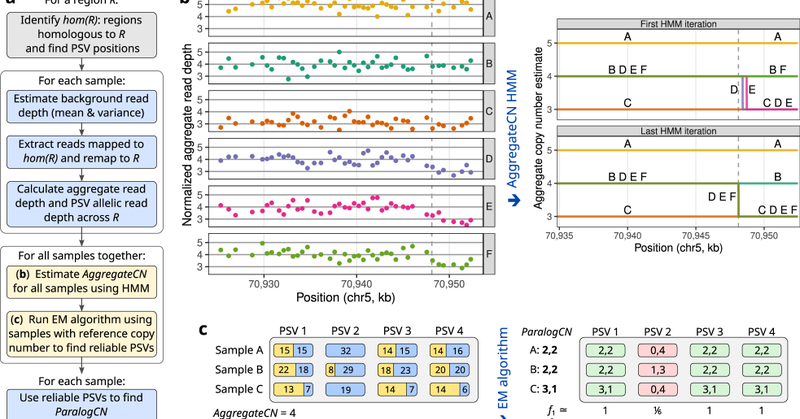
To fix that, we run EM-algorithm to jointly find a subset of reliable PSVs and paralog-specific copy numbers for all samples. We require many samples to find the set of reliable PSVs. However, one can detect CNVs for a single sample if the region was analyzed previously. [6/7]

1
0
0
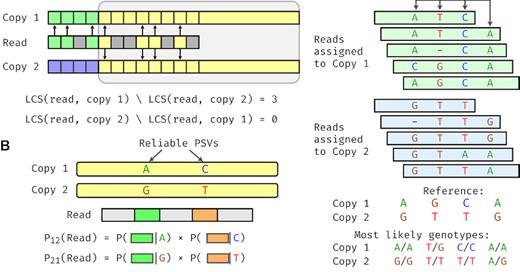
We use paralogous sequence variants (PSVs) — small differences between repeat copies — to find paralog-specific CN. Unfortunately, many PSVs are unreliable (same allele on several repeat copies), the fact that is often ignored when analyzing duplicated regions. [5/7].
1
0
0
To find aggregate CN, we use a Hidden Markov Model. Observations: read depth in consecutive 100 bp windows. Hidden states: aggregate CN values. We refine model parameters based on multiple samples to find deletions and duplications more accurately. [4/7]

1
0
0
As input, Parascopy takes single or multiple short-read WGS BAM/CRAM files and a list of target low-copy repeats. For each region, Parascopy outputs aggregate copy number (total CN across all repeat copies) and paralog-specific CN (CN for each individual repeat copy). [3/7]

1
0
0
Low-copy repeats, also known as segmental duplications, cover up to 5% of the genome, are prone to extensive copy number variation and are notoriously hard to analyze. In this paper, we present a novel computational method, Parascopy [2/7].
1
0
0
I am delighted to share our new paper in @NatureComms on identifying copy number for duplicated genes! #bioinformatics #genomics #structuralvariation. A short thread below: [1/7].
nature.com
Nature Communications - Low-copy repeats cover up to 5% of the human genome and are prone to extensive copy number variation. Here, the authors present a novel computational method to estimate...
2
2
10
RT @HiTSeq: Copy number discussions continue with @TimofeyProdanov: "Robust and accurate estimation of paralog-specific copy number for dup….
0
1
0
Check out our paper on mapping long reads to segmental duplications:
academic.oup.com
Abstract. The ability to characterize repetitive regions of the human genome is limited by the read lengths of short-read sequencing technologies. Although
0
0
2