
Roland Faure
@rfaure2
Followers
21
Following
70
Media
4
Statuses
30
Ph.D. student in genome assembly and phasing, GenScale team in Rennes
Joined May 2021

FroM Superstring to Indexing: a space-efficient index for unconstrained k-mer sets using the Masked Burrows-Wheeler Transform (MBWT) https://t.co/YagAm5YBz3
#biorxiv_bioinfo
0
10
32

Today we’re excited to freely share an early-version of, perhaps, the world’s most expansive genetics dataset: Logan. #bioinformatics #petabase #genetics #genomics #openscience
https://t.co/CQSBmvn7se

biorxiv.org
The NCBI Sequence Read Archive (SRA) is the largest public repository of DNA sequencing data, containing the most comprehensive snapshot of Earth’s genetic diversity to date. As its size exceeds 50.0...
6
139
320

A new nanopore sequencer from BGI https://t.co/mnz3LdErL1 Unfortunately, they do not provide reads to evaluate them.

biorxiv.org
Nanopore sequencing, a third-generation sequencing technology, has revolutionized the gene sequencing industry with its advantages of long reads, fast speed, real-time sequencing and analysis, and...
0
10
19
In what journal do you publish? I am looking for well-established bioinformatics journals with reasonable APC, but I can't find anything below $3000 😓
0
0
0
Soon, we will see more long and ultra-long read technologies that combine sequencing, sequence alignment and AI for error correction. I am proud that our lab has contributed to this by https://t.co/hHFbezdGeJ
https://t.co/MjKazNVExU and https://t.co/KyR3LPx5rT,
github.com
HERRO is a highly-accurate, haplotype-aware, deep-learning tool for error correction of Nanopore R10.4.1 or R9.4.1 reads (read length of >= 10 kbps is recommended). - lbcb-sci/herro
1
21
97

[1/11] Thrilled to announce our latest preprint on function-assigned masked superstrings (shortly, f-masked superstrings or f-MS), which transform masked superstrings into an operationally complete data type for k-mer sets. w/@sladky_on and @VeselyPavel_mff

Function-Assigned Masked Superstrings as a Versatile and Compact Data Type for 𝑘-Mer Sets https://t.co/vEVldKLJzH
#bioRxiv
3
12
23
Very happy to receive the best student's paper award at the Bioinformatics part of BIOSTEC2024 conference 🥳! Work on using ILP to improve metagenomic assembly. Soon on biorXiv. With Rumen Andonov and Tam Truong in the @GenscaleTeam.
1
0
1
Could this be useful in genomics? Two reasons to hope 1) When divergence is low the alignment is near-optimal 2) O(n) in time and O(1) in space => probably very fast if well-implemented 🧵6/6
0
0
0
Switched from random walk to methodic exploration of the matrix: overestimation comes down to O(s)! (but this cannot be formalized as an embedding anymore) 🧵5/6
1
0
0
Is that good in practice -> I tested -> NO. Random walks are very slow to explore the space. But what if we got rid of the randomness ? 🧵4/6
1
0
0
CGK alignment will eventually "find" the optimal alignment after a non-matching character with a random walk. If s is the divergence, the overestimation of the distance is in "O(s^2) with probability 2/3" 🧵3/6
1
0
0
The translation of CGK to pairwise alignment is simple: greedily accept matches between sequences, and if two characters don't match... randomly insert/delete/mismatch! This is what it looks like on NW matrix 🧵2/6
1
0
0
Just discovered CGK embedding by @KoudyMK lab. Very fun paper: aligning two sequences in O(n) time and O(1) space 😯 sacrificing a bounded amount of precision. https://t.co/gMlwdzybMj Summary of the journal club in @GenscaleTeam 🧵1/6
1
0
2
HairSplitter can recover more strains compared to previous methods, while being faster. 🧵 3/3
0
0
1
General purpose long-read assemblers like metaFlye generally collapse close strains. The methods available for now to recover the strains were Strainberry, from @r_vicedomini and @RayanChikhi, and iGDA, but worked only for simple metagenomes 🧵2/3
1
0
1
Want strain-specific metagenomic assemblies from Nanopore reads ? HairSplitter is on bioRxiv https://t.co/C3mA4yYHue 🧵1/3
biorxiv.org
Motivation Long-read assemblers face challenges in discerning closely related viral or bacterial strains, often collapsing similar strains in a single sequence. This limitation has been hampering...
2
3
7

1) Assemblers do not like structural heterogeneity! Often results in a misassembly. 2) Some misassemblies are completely mysterious and have no clear cause. 3) Assembly can be non-deterministic: re-running an assembly with identical parameters can give a different result. (2/2)
1
1
13
QuickDeconvolution is out! Very happy to have developed that within the @GenscaleTeam. Deconvolve your linked reads data to finally have one cloud per barcode.
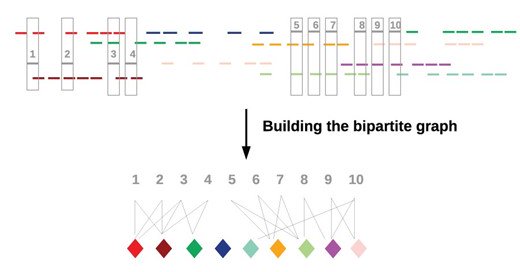
academic.oup.com
AbstractMotivation. Recently introduced, linked-read technologies, such as the 10× chromium system, use microfluidics to tag multiple short reads from the
0
0
1

🎥 Il reste de la place pour le festival Sciences en Cour[t]s ! 🗓️ 13 octobre 🗺️ Diapason ⌚️ 20h 🎫 https://t.co/EXKWb4M7CS 👋 @DiapasonRennes @UnivRennes1 @UnivRennes_2 @Espace_sciences @FeteScience @FondationR1 @IGDRennes @metropolerennes @GROUPEESRA
0
6
3