

Wouter Saelens
@Zouters
Followers
189
Following
997
Media
3
Statuses
98
Post-doc at Bart Deplancke lab @ EPFL
Lausanne, Switzerland
Joined March 2016
Still, there's much left to explore, as we "just" implemented two models here. We're working on integrating the sequence directly, and also thinking about interfacing with time dynamics tools.

biorxiv.org
Machine learning methods that fully exploit the dual modality of single-cell RNA+ATAC-seq techniques are still lacking. Here, we developed ChromatinHD, a pair of models that uses the raw accessibil...
0
0
0
ChromatinHD is available at . It's still in beta, but you can already run the main models and interpretation tools. The full-featured package, interfacing with e.g. SCENIC+ @steinaerts, is coming soon. We're happy to hear any feedback.

github.com
High-definition modeling of chromatin + transcriptomics data - DeplanckeLab/ChromatinHD
1
0
0
Why? We cross-referenced with ChIP-seq data and found that these large fragments are indicative of very dense binding of TFs. The model learned independently that these fragments are associated with higher expression.
1
0
0
Our model also learns the importance of fragment size. Beyond the expected nucleosomes, our model also learned that large fragments just below the nucleosomal size (60-120bp) are much more predictive than fragments that straddle 10-60bp, the typical size of a TF footprint.
1
0
0
What is intriguing is that if we cross-reference with DNA contact data (Hi-C), we find that DNA contact and co-predictivity is juxtaposed (1-5kb) in such a way as to "present" the co-predictive regions to the outside of a loop. Hub formation right before our eyes!

1
0
0
We first define co-predictivity as two position that, if they are both open, predict higher expression than the two positions individually. Being defined based on accessibility, this feature is similar but complementary to DNA contact or proximity data.

1
0
0
This is of course just at the level of genomic positions, but our models can capture much more. .
1
0
0
It's really fascinating to look at the this at the gene-level, because almost every gene has some special predictive/differential accessibility changes going on that are not easily captured by peak/window-based methods.

1
0
0
To make the more complex models interpretable, we implemented several interpretation tools. For example, we can interpret at the genomic position and then define "peaks" (that often do not look like peaks) that are predictive or differential.
1
0
0
Through comprehensive benchmarking, we first show that our models are better in predicting gene expression and capture more functional TF binding or genomic variation.
1
0
0
We can learn these dependencies in a purely data-driven way. We constructed two models: ChromatinHD-pred will predict gene expression from raw fragments. ChromatinHD-diff learns differential accessibility between cell types/states using raw fragments.
1
0
0
We hypothesized that ATAC-seq data contains information that goes beyond counts of peaks or windows. What if the relevant resolution depends on the position? Or if fragment size or other fragments within the same cell contain important information?.
1
0
0
Ever looked at your ATAC-seq signal and thought: there must be a better way to model this than peaks or windows? Happy to present ChromatinHD, a method that models scATAC+RNA data using the raw fragments. With @BartDeplancke @OlgaPushkarev
biorxiv.org
Machine learning methods that fully exploit the dual modality of single-cell RNA+ATAC-seq techniques are still lacking. Here, we developed ChromatinHD, a pair of models that uses the raw accessibil...
4
33
126
RT @FreyaRSvedberg: The @NatImmunol perspective @MartinGuilliams and I wrote is out. We ask does tissue imprinting restrict macrophage plas….
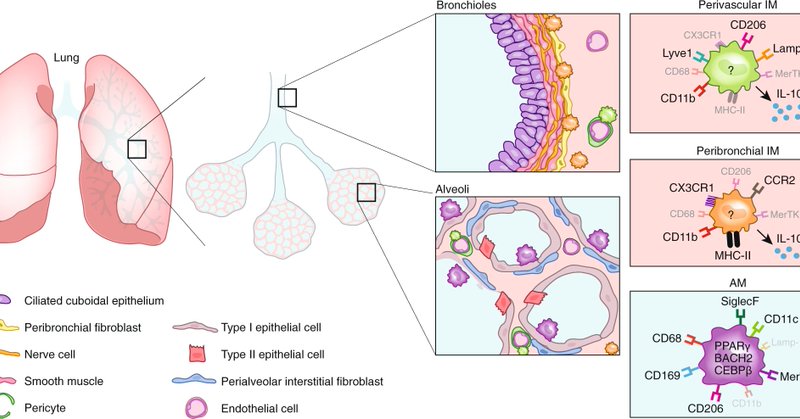
nature.com
Nature Immunology - Based on the results of recent studies that have dissected the response of individual macrophage subsets to pulmonary insults, Guilliams and Svedberg call for an adjustment of...
0
66
0
RT @NatureBiotech: Generalizing RNA velocity to transient cell states through dynamical modeling .
0
127
0
0
2
0
RT @cziscience: #Opensource software is essential to science. That’s why we’re awarding 23 grants to support open source tools that acceler….

chanzuckerberg.com
CZI supports software projects that accelerate biomedical research
0
98
0
RT @danwagnerlab: With widespread adoption of single-cell-omics technologies, developmental biologists are increasingly relying on the use….
0
165
0
RT @koenvdberge_Be: Our work on differential expression (DE) analysis downstream of trajectory inference (TI) is now published! A small thr….
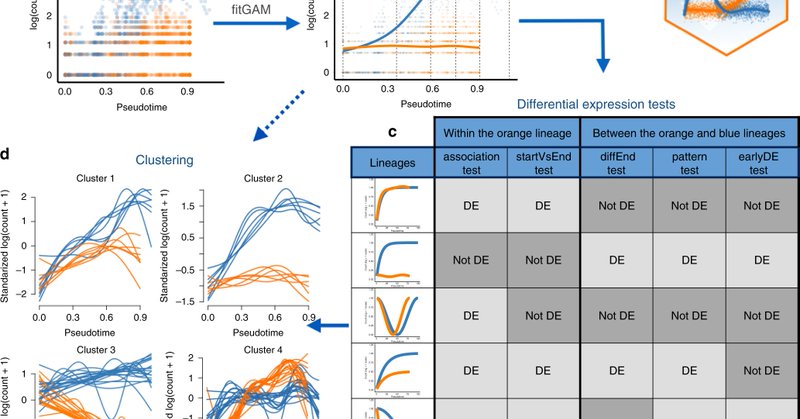
nature.com
Nature Communications - Downstream of trajectory inference for cell lineages based on scRNA-seq data, differential expression analysis yields insight into biological processes. Here, Van den Berge...
0
39
0
RT @fabian_theis: We're excited to release a #scanpy update for the analysis and visualization of #spatialTranscriptomics data, focussing o….
0
107
0