

Teddy Koker
@teddykoker
Followers
869
Following
535
Media
26
Statuses
125
PhD student at @MIT; machine learning for physics.
Cambridge, MA
Joined March 2018
New pre-print! With @keeganquigley, @etaw_ucb, Kevin Tibbetts, and Lin Li at @MITLL. We introduce ChargE3Net, a higher-order equivariant neural network for predicting electron density in materials. Paper: Code:

1
1
7
RT @BigAmeya: Really excited to (finally) share the updated JAMUN preprint and codebase! We perform Langevin molecular dynamics in a smooth….
0
5
0

Generate videos in just a few seconds. Try Grok Imagine, free for a limited time.
385
673
3K
RT @vdbergrianne: 🚀 After two+ years of intense research, we’re thrilled to introduce Skala — a scalable deep learning density functional t….
0
61
0
With slight changes of the initialization and hyperparameters, we achieve improved RMSE on the 3BPA test set! This work serves as a simple introduction to training and testing equivariant neural network potentials in JAX, and can be easily extended for new methodologies. n/n

0
0
0
In order to verify correctness of the implementation, we compare performance on the 3BPA dataset to two different PyTorch NequIP implementations: (1) @simonbatzner et al.'s "nequip" repo, and (2) @IlyesBatatia et al.'s "mace" repo. 3/n.
1
0
0
The package includes code for training and evaluation of neural network potentials, a calculator for usage with Atomic Simulation Environment (ASE), and pre-trained weights for the 3BPA dataset. 2/n.
1
0
0
Introducing nequip-eqx, a JAX implementation of the popular NequIP interatomic potential model. Repo: The goal of the repository is to offer a simple (<1000 lines of code) implementation while providing competitive performance to existing codebases. 1/n
1
1
4
RT @n_gao96: I am truly excited to share our latest work with @MScherbela, @GrohsPhilipp, and @guennemann on "Accurate Ab-initio Neural-net….

arxiv.org
We present finite-range embeddings (FiRE), a novel wave function ansatz for accurate large-scale ab-initio electronic structure calculations. Compared to contemporary neural-network wave...
0
32
0
RT @TacoCohen: Does equivariance matter at scale?. When the twitter discourse gets so tiring that you actually go out and collect EVIDE….
0
53
0
RT @pfau: I’m beyond thrilled to share that our work on using deep learning to compute excited states of molecules is out today in @Science….
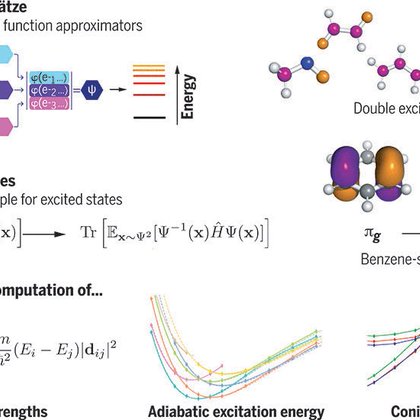
science.org
We present an algorithm to estimate the excited states of a quantum system by variational Monte Carlo, which has no free parameters and requires no orthogonalization of the states, instead transfor...
0
171
0
The only operation implemented thus far is the tensor product - a core operation within the framework. After several optimizations and leveraging of C code generation, we can achieve faster CPU performance for tensor products than the JAX and PyTorch versions at varying max l.

0
0
6
Introducing e3nn.c: . A pure C partial implementation of e3nn, a modular framework for neural networks with Euclidean symmetry. Created for pedagogical purposes, but similar code could be used for full C/C++/CUDA implementations of e3nn models.

1
3
10
RT @xiangfu_ml: Charge density is the core attribute of atomic systems in DFT. When representing and predicting charge density with ML, it….
0
47
0
RT @TimothyDuignan: Another very impressive general purpose graph NN for molecular simulation. Feels like this field is really accelerating….
0
37
0
As electron density is fundamental to predicting material properties, we view models that compute electron densities as a potential intermediate towards other material properties that may improve the generalization of machine learning models in material.science!.
0
0
0
Lastly, we use ChargE3Net densities within DFT non-self-consistently, demonstrating its ability reproduce several physical properties such as energy, forces, band energies, and band gaps.

1
0
0
Furthermore, ChargE3Net densities can be used to initialize DFT, greatly reducing the number of self-consistent field (SCF) steps required for DFT to converge to a solution.

1
0
0
Unlike traditional DFT methods, our model scales linearly with respect to the number of atoms in the system, enabling computation of electron density on larger systems than previously feasible.

1
0
1
We attribute improved accuracy to the introduction of higher rotation order features within the network, which are necessary for effectively modelling systems with high angular variation in electron density, such as those with covalent bonding.

1
0
1
Evaluated against DFT-computed charge densities, ChargE3Net demonstrates an improved normalized mean absolute error on large and diverse datasets of materials (Materials Project/MP) and molecules (QM9) compared to prior work.

1
0
0
The calculation of electron density distributions using density functional theory (DFT) in materials and molecules is central to the study of their quantum and macro-scale properties, yet accurate and efficient.calculation remains a long-standing challenge.
1
0
0