

Kyle Tretina, Ph.D.
@AllThingsApx
Followers
4K
Following
16K
Media
632
Statuses
8K
Follow for AI in Digital Biology and Drug Discovery @NVIDIA, ex Insilico Medicine, ex Yale, PhD UMaryland, views are mine, DM for collabs
Boston, MA
Joined November 2013
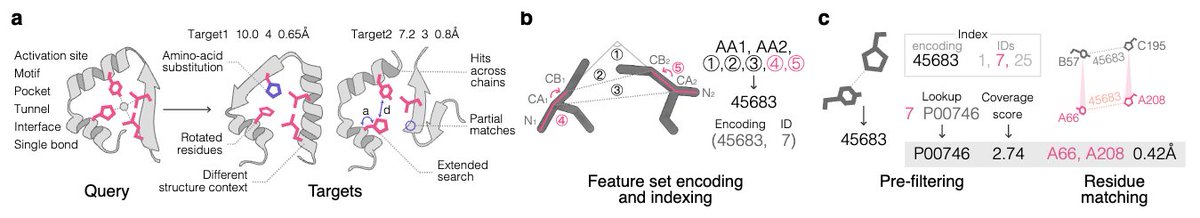
🧬Motifs are the new BLAST. Folddisco scans 53M AlphaFold structures for catalytic, binding or allosteric patterns in seconds🔎. 18× quicker, 3.5× smaller, hits in seconds🌌
5
50
348
RT @genophoria: We put a blog post together to tell you about all that went behind the scenes for @arcinstitute's Virtual Cell Challenge: (….
0
32
0

RT @gdb: Our custom LLM, gpt-4b micro, has helped achieve an advance in biology. It designed novel variants of the Nobel-winning Yamanaka….
0
173
0
RT @ikalvet: RFdiffusion2 is now live!. You can now design proteins, and in particular enzymes from just partially….
0
95
0
RT @EpochAIResearch: What's the best model you can run on a single consumer GPU?. We've updated last week's Data Insight with 3 additional….
0
26
0
RT @NikoMcCarty: I'm (slowly) writing the book I've been thinking about for the last 3+ years. Nothing official yet, but I'm hoping to wri….
0
148
0
A new self driving lab out of NC State University:.
sciencedaily.com
A new leap in lab automation is shaking up how scientists discover materials. By switching from slow, traditional methods to real-time, dynamic chemical experiments, researchers have created a...
0
0
1
I'd love to hear more chatter on X about this paper. Is this a promising approach?.
0
0
0
Read the preprint:. Quantum-Boosted High-Fidelity Deep Learning.

arxiv.org
A fundamental limitation of probabilistic deep learning is its predominant reliance on Gaussian priors. This simplistic assumption prevents models from accurately capturing the complex,...
1
1
1
Gaussian priors are holding back bio‑AI. QBM‑VAE uses a Boltzmann prior sampled on an Ising QPU. On 1M+ single cells it beats scVI on integration, rare cells & trajectories. I didn't have "quantum AI meets single-cell" on my BINGO card for this month.

2
7
39
Low‑Tc ≠ truly novel. Subtle substitutions can push Tc below 0.4 yet retain the same core scaffold, and even when scaffolds differ, binding pharmacophores are often conserved. I guess novelty should be assessed beyond 2D fingerprints.

0
0
0
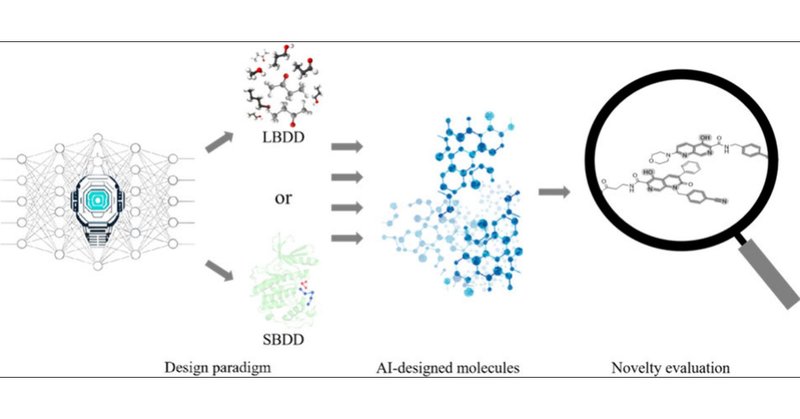
The paper:. AI-Designed Molecules in Drug Discovery, Structural Novelty Evaluation, and Implications.
pubs.acs.org
Achieving structural novelty in drug discovery remains a critical challenge. Artificial intelligence (AI) has demonstrated remarkable potential in deciphering the complex relationships between...
1
0
0
🥶AIDD still struggles to explore new chemical space (ice cold take?)🧊. In 71 studies, Ligand-Based Drug Design hits often echoed known actives (Tcmax>0.4 in 58%), but Structure-Based Drug Design fared better (18%). How would you measure molecular novelty?

1
0
1
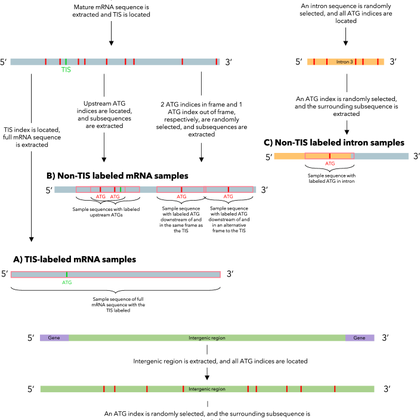
Paper:. NetStart 2.0: prediction of eukaryotic translation initiation sites using a protein language model.
bmcbioinformatics.biomedcentral.com
Background Accurate identification of translation initiation sites is essential for the proper translation of mRNA into functional proteins. In eukaryotes, the choice of the translation initiation...
0
0
3
“Protein‑ness” beats Kozak. NetStart2.0 translates ±100aa, asks ESM‑2 “does this look like a protein?”, and picks the right start codon = SOTA across 60 species. Next up: pLMs for splice sites & stops? 🔬🧬

1
1
7
The LBVS accuracy jump comes mostly from the new energy potential, not from search. Cross‑rescoring shows that AutoDock‑SS poses become strong when scored with the LJ‑like potential, while UniDock‑Pro poses collapse under the old recursive model.

0
0
2
Paper:. UniDock-Pro: A Unified GPU-Accelerated Platform for High-Throughput Structure-Based, Ligand-Based, and Synergistic Hybrid Virtual Screening.
1
1
2
UniDock‑Pro blends protein grids + ligand‑similarity maps and screens millions/day. 🔥6.4M lig/day on one RTX 4090.🎯LBVS EF1% ↑ 2.45× vs AutoDock‑SS.💁FFCA tells you when Hybrid helps. == GPU acceleration + scoring‑function engineering

1
3
30
RT @PossuHuangLab: We have a new collection of protein structure generative models which we call Protpardelle-1c. It builds on the original….
0
28
0