
Vanja (Ivan Kulakovskiy)
@halfacrocodile
Followers
198
Following
348
Media
52
Statuses
137
bald but bearded bioinformatics buddy
Joined March 2022
(1/4) Thrilled to announce another major release of the HOCOMOCO motif collection, well-known for its silly name and rigorous approach to constructing and benchmarking DNA sequence motifs recognized by human and mouse transcription factors.

1
11
55
RT @GallowayLabMIT: So you want to change transgene expression: just change your promoter, right? Changing the promoter increases RNA and t….
0
115
0
RT @febos41: 📢Job Alert: Postdoctoral Researcher in the Laboratory of RNA Algorithms @IMol_Institute, up to three years, fully funded by @N….
0
1
0
RT @molina_lab: 🚨 New method alert!.We developed HiddenFoot, a physics-inspired approach to decode single-molecule footprinting data and in….
0
7
0
(15/15) MIXALIME is written in Python, getting the stable version is as easy as 'pip install mixalime', and the source code plus tutorial are freely available at

0
0
1
(14/15) We hope you will find MIXALIME and UDACHA useful in your study of regulatory sequence variants.
1
0
1
(13/15) According to stratified LD score regression, these rSNPs correspond to regulatory regions involved in cell type-specific phenotypes. Most importantly, the collection of significant rSNPs can be fully explored at

1
0
1
(12/15) AS-chromatin variants are predominantly located in promoter and enhancer regions and significantly overlap ADASTRA ASBs and GTEx eQTLs.

1
0
1
(11/15) Finally, we used MIXALIME to analyze 5858 chromatin accessibility datasets from In the end, we identified >200 thousand allele-specific chromatin accessibility variants.

1
0
1
(10/15) In most cases, MIXALIME outperforms other popular tools for AS calling, offering a good sensitivity/specificity tradeoff.

1
0
1
(9/15) Using heart CAGE-Seq data from Deviatiiarov et al., we benchmarked MIXALIME by calling AS variants in CAGE-Seq and comparing them to allele-specific transcription factor binding from ADASTRA and eQTLs from GTEx.
1
0
1
(8/15) Copy-number variation and aneuploidy are accounted for by fitting a mixture model assuming that reads originate from haplotypes with different copy numbers.

1
0
1
(7/15) MIXALIME also handles reference mapping bias and aneuploidy, see the underlying math in To counter the mapping bias, MIXALIME uses separate fits for Alt read counts with the fixed number of Ref reads and vice versa.

1
0
1
(6/15) MIXALIME provides a variety of statistical models to fulfill particular use cases, from a standard binomial model to the beta negative binomial (BetaNB) model that accounts for extra overdispersion.

1
0
1
(5/15) MIXALIME is a novel toolbox that uses MIXture models for ALlelic IMbalance Estimation. In the paper, we describe a general workflow from FASTQ files to allelic read counts and SNV-level allele-specific statistics.

1
0
1
(4/15) Technically, the allele specificity is revealed by counting the number of reads supporting each of the alleles and estimating the statistical significance of the observed allelic imbalance.

1
0
1
(3/15) High-throughput sequencing allows tracking chromatin state, gene expression, protein-DNA interactions, and more. Eventually, all methods yield short reads that can be used to call single-nucleotide variants and assess the allele specificity.

1
0
1
(2/15) Check the original Tweetorial describing the respective preprint or follow the thread below.
We present MIXALIME, a versatile software for assessing allelic imbalance in sequencing data with mixture models, that we used to build UDACHA, Uniform Database of Allele-specific CHromatin Accessibility in the human genome, see (1/14)

1
0
1
(1/15) Yet another sweet bioinformatics "software+database" couple from our team:.Meet MIXALIME, a framework for assessing allelic imbalance, and UDACHA, a database of allele-specific chromatin accessibility, read more at

1
6
18
Last but not least: this update became possible thanks to the experimental data & motif analysis performed within the Codebook/GRECO-BIT collaboration,
0
1
0
(4/4) Don't hesitate to grab a fresh release from and remember that we also provide a fancy online motif scanner, MoLoTool, in all its interactive JS-powered beauty.
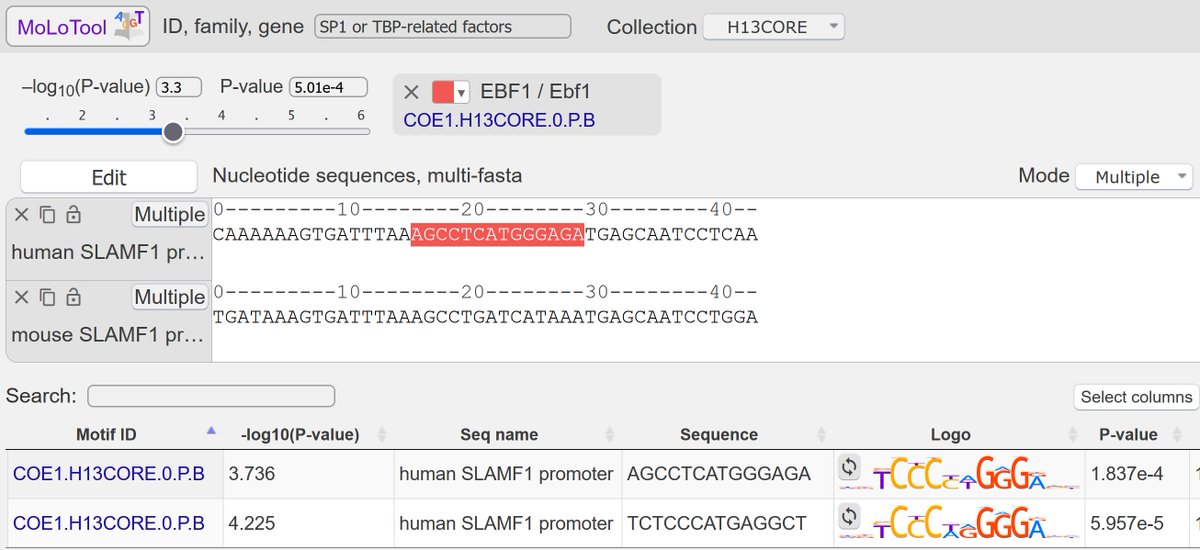
1
1
0