

Jimmie Ye
@yimmieg
Followers
3K
Following
45
Media
57
Statuses
278
Professor @ UCSF, Genomicist, Geneticist, Adopted Immunologist, #GirlDad, Immigrant
San Francisco, CA
Joined June 2012


15/🧵Dive deeper into our methodology and findings in the full paper, where we explain how Memento can empower your single-cell analyses and drive new insights into gene regulation. Also, Memento is compatible with scanpy and can be accessed here:
0
1
5
14/🧵Biology is fundamentally quantitative, and advancing the field requires robust mathematical and statistical frameworks. Working on Memento with Mincheol deepened my appreciation for these principles, and we hope to see more rigorous approaches to scRNA-seq analysis.
1
0
1
13/🧵This thread captures years of dedicated effort by Mincheol, who addressed one of the core challenges in scRNA-seq analysis. I highlighted a few of many papers that inspired us. Ultimately, methods matter—and the people behind those methods matter even more.
1
0
0
12/🧵One more thing: Memento’s shuffling strategy can be precomputed, allowing for deployment on massive datasets like @cziscience's 50 million cell atlas. We showcased its utility with the @cellxgene Discover API, enabling near-instant comparisons across diverse cell groups.

1
0
1
11/🧵In all cases, Memento outperformed existing methods by identifying more significant and reproducible changes in mean gene expression. It also uncovered new modes of transcriptional regulation by analyzing changes in expression variability and gene correlation.
1
0
1
10/🧵Beyond mean expression, Memento can also be used to perform differential variability (DV) and correlation (DC) analysis. We demonstrated its utility in three contexts: epithelial cells responding to interferon, Perturb-seq analysis of T cells, QTL mapping in 1.2M PBMCs.

1
0
0
9/🧵Key result: Memento enables differential mean expression analysis (DM) that outperforms pseudobulk, addressing observations by Squair et al. (. We validated Memento by comparing DM results from sample matched bulk RNA-seq and scRNA-seq data.

1
1
5
8/🧵MoM estimation necessitates computation of confidence intervals for parameter comparisons. Mincheol developed a bootstrapping approach that exploits the logarithmic growth of unique counts with cell number in scRNA-seq data, allowing efficient resampling in large datasets.
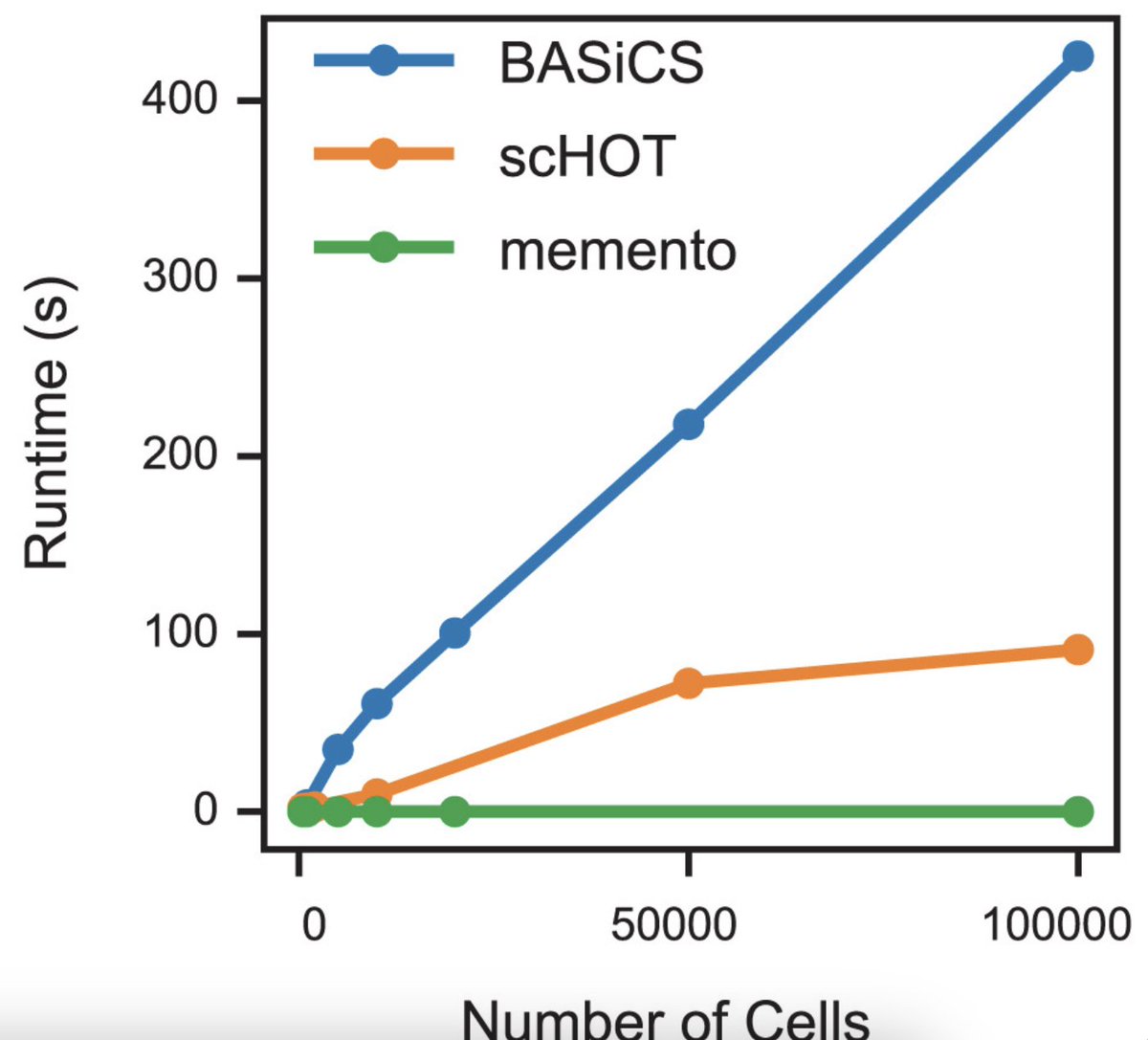
1
0
0
7/🧵In Memento, we leverage method-of-moments (MoM) estimators for the mean, residual variance, and gene correlation from scRNA-seq data. Memento’s estimates are better correlated with smFISH data generated by @arjunrajlab ( than imputation-based methods.
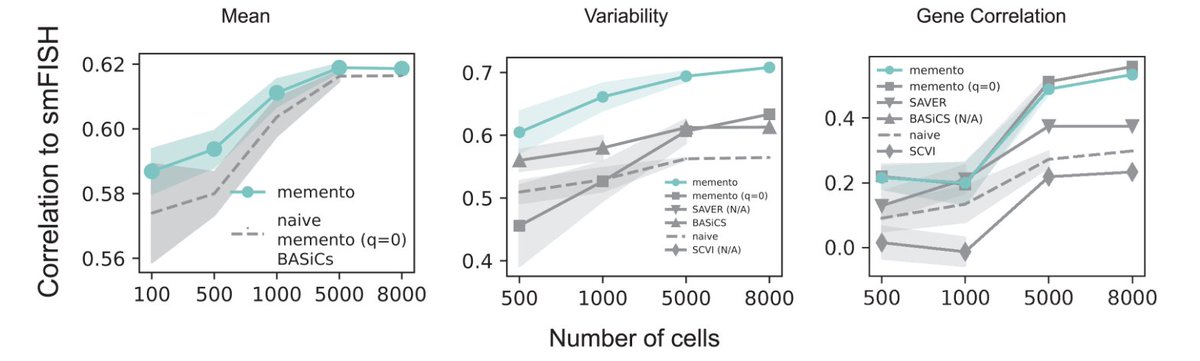
1
0
1
6/🧵Why is modeling the sampling process of scRNA-seq important? As articulated by Sarkar et al. (, separating biological from technical noise is critical for downstream quantitative analysis.
1
0
2
5/🧵We were inspired by @KleinLabHMS's InDrop paper that used the hypergeometric to model noise in scRNA-seq ( S19) and Zhang et al. that used a Poisson approximation (.
1
0
0
4/🧵Mincheol Kim, an MD/PhD student in the lab, wondered if its possible to develop a rigorous model of the scRNA-seq sampling process. He provided the first quantitative demonstration that scRNA-seq can be modeled as a hypergeometric sampling process.
1
0
1
3/🧵@MattG_Jones initially experimented with a “minipseudobulk” approach to improve variance estimates. Our strategy was very similar to the MetaCell framework from Baran et al. However, minipseudobulk was a heuristic, not a solution.
1
1
2
2/🧵Our journey began ~7 years ago, grappling with the challenges of data sparsity in scRNA-seq data, particularly for accurately estimating the cell-to-cell variances of individual genes, a key parameter for understanding transcriptional regulation and cellular heterogeneity.
1
0
1
1/🧵Excited to re-introduce Memento in (, a scalable method for differential analysis of scRNA-seq data. It provides first-in-class performance in statistical power and calibration for comparing differences in mean, variability, and gene correlation.👇.
3
61
277
RT @J_E_Mitchel: It's great to see my first paper of the PhD published in @NatureBiotech! This describes our scRNA….
0
48
0
RT @GladstoneInst: 🚨 NEW PUBLICATION 🚨 "Systematic decoding of cis gene regulation defines context-dependent control of the multi-gene cost….
0
6
0
Ugh pretty annoyed with the high frequency of thefts on UCSF campus. Someone somehow got into our locked space this morning and took a bunch of stuff (headphones, laptop, cough drops). But this…really?

5
1
32