

Daan de Groot
@daandegroot91
Followers
82
Following
288
Media
7
Statuses
84
Postdoc in mathematical biology; Run-Bike-Run-fanatic. Interested in all systems that are complex enough
Joined July 2009
"How can 'simple' microbes adapt to so many different environments, even when these environments change unpredictably?" This is the question that @TjalmaAge and me set out to answer in 2019. Our main finding was just published: A short background story:.

pnas.org
Microbes in the wild face highly variable and unpredictable environments and are naturally selected for their average growth rate across environmen...
1
12
35
RT @NimwegenLab: Here it is! Bonsai. No more excuse to use t-SNE/UMAP. Bonsai not only makes cool pictures of your data. It actually rigoro….

biorxiv.org
Single-cell omics methods promise to revolutionize our understanding of gene regulatory processes during cell differentiation, but analysis of such data continues to pose a major challenge. Apart...
0
5
0
RT @NimwegenLab: Very exciting new masters program at the Biozentrum that I look forward teaching in together with colleagues like @Zavolan….
0
10
0
RT @NimwegenLab: Dear scRNA-seq people. I'd like the hive's opinion. What current scRNA-seq datasets or dataset collections would you say a….
0
7
0
RT @NimwegenLab: The Swiss science foundation SNF has been functioning superbly and is the envy of many abroad. Now there are VERY worrisom….

secure.avaaz.org
Dear President, dear members of the Foundation Council of the SNSF,We, the undersigned, have been alarmed to learn of the substantive changes that have been planned for the future functioning of the...
0
19
0
RT @RJvTatenhovePel: We are hiring! Are you looking for a Postdoc, excited about applying micro-compartmented cultivation systems in indust….
careers.tudelft.nl
0
17
0
RT @sees_lab: 📢 New in @NatureComms we investigate the limits of symbolic regression: Can we always learn a model from data? We find a tran….

nature.com
Nature Communications - Learning analytical models from noisy data remains challenging and depends essentially on the noise level. The authors analyze the transition of the model-learning problem...
0
49
0
metabolites and reactions). Hopefully, at some point, ECM enumeration will become a standard step in any metabolic reconstruction pipeline, such that a full metabolic characterization of any sequenced organism is always available.
0
0
2
in which ecmtool may be scaled even further. We hope that publishing this improved version will motivate more people to make it better! Because we are getting closer and closer to being able to enumerate all ECMs for any genome-scale metabolic network (which typically have ~2000.
1
0
1
As a consequence, we could now scale ECM-enumeration further than ever before, such that we could calculate all ECMs for a synthetic minimal cell with over 300 metabolites and reactions using 60 cores! Is ecmtool now completely done and perfect? Well no, there might still be ways.
1
0
1
advantages for ECM-enumeration: 1) LRS does not produce enormous amounts of intermediate candidates, 2) extreme rays that you already passed on your reverse search can be written into an output file and do not have to be kept in working memory, and 3) it can be done in parallel!.
1
0
1
However, this problem was explored by Avis et al., (2017) and they found a way of parallelizing this reverse search efficiently such that its speed scales almost linearly with the number of computation cores. Using this “mplrs”-tool provides three clear.
1
0
1
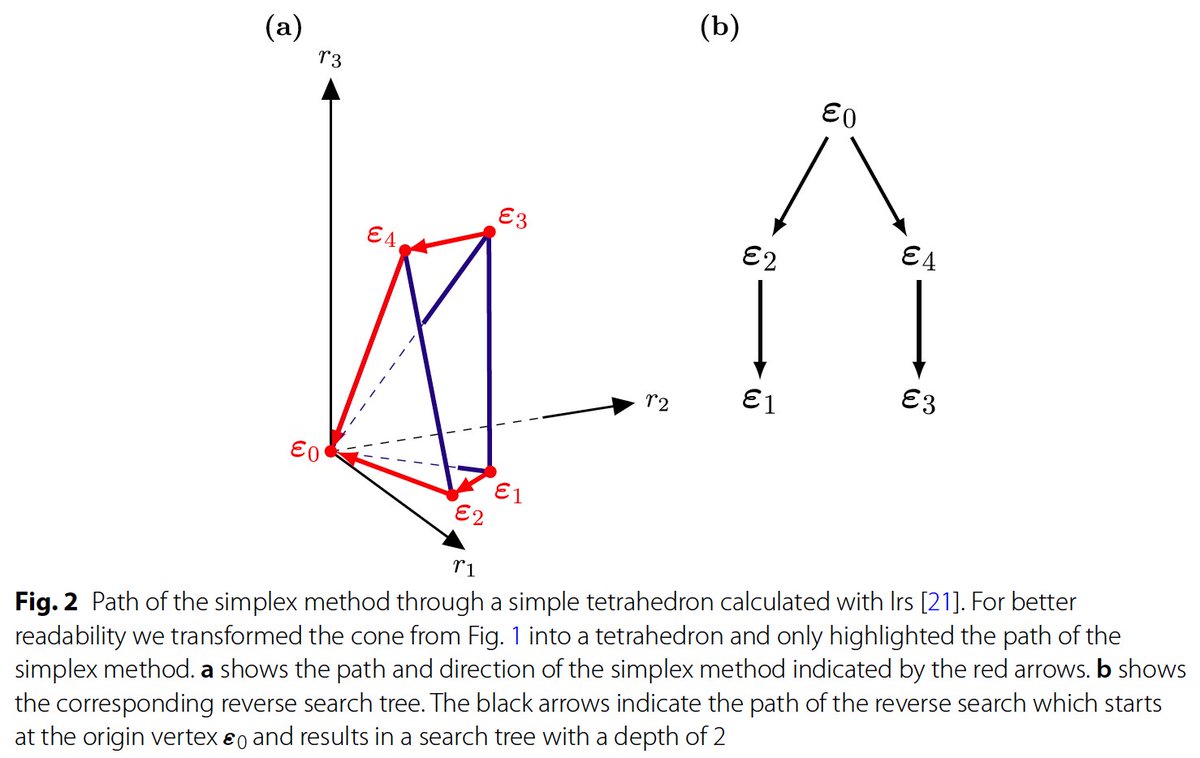
the simplex method could have taken. When it goes down all paths, it must have found all possible vertices of the cone! Now this sounds very inefficient!.
1
0
1
We are thus at the optimum of some linear-programming problem. Usually, one can find such optima by following the “simplex method” which walks from vertex to vertex until it has found the best one. LRS reverses this: it starts rom this optimum, and follows all possible paths that.
1
0
1
Lexicographic Reverse Search (LRS): . This method does a much more straightforward thing than the DD-method. It starts on vertex of the chopped-off final cone (equivalent to a ray of the cone) and asks for a linear objective which is optimized there.
1
0
1
before it can select the minimal set with which it can move on. In a genome-scale metabolic network there can really be so many candidates that you cannot store them anymore! Now @BeeAnka_A and @enit7677 had the idea to use a different ray-enumeration algorithm called.
1
0
1
that you could make from e1 and e4. Once we selected the non-redundant rays, we have a set of candidate rays that all satisfy a x >= 0. Now we can go to the next linear inequality b x >= 0, and so on. However, the DD-method needs to store all these intermediate candidates.
1
0
1
want to keep. There are typically many many of these candidates, but you only want to keep the 'extreme' ones to obtain a minimal set of ECMs, and the DD-method does that for you. In figure (b) above, you want to keep e6 that you could make from e1 and e5, but not the candidate.
1
0
1
Each of your candidates, c, will either be “-“: a c < 0, “0”: a c = 0, or “+” a c > 0. Candidates of the latter two types are fine, but the “-“candidates should be dropped. However, you can combine these “-“ candidates with “+”candidates to make new “0”candidates that you then.
1
0
1
the Double Description method to solve it. I’m using a Figure from to explain it. Basically, you start from a set of candidate rays that does not satisfy any of the linear inequalities yet, and then you add one of the linear inequalities: a x >= 0.

1
0
1
To calculate the ECMs you need to do the following computation twice: given a convex polyhedral cone, defined as the subspace satisfying a certain set of linear inequalities (Ax >= 0), find all of its “extreme rays”. This is a known computational problem and in ecmtool we used.
1
0
1