
Q-Chem
@QChemSoftware
Followers
2K
Following
334
Media
684
Statuses
1K
Q-Chem provides a comprehensive ab initio quantum chemistry program, allowing scientists worldwide to solve computational problems quickly and accurately.
Pleasanton, CA USA
Joined January 2012
Q-Chem 6.3 is here! Upgrade to enjoy improved performance and usability, and take advantage of new tools for studying chemistry and spectroscopy. Read about new features: Interested in trying it out yourself? Request a free trial:

0
0
11
Congratulations to Q-Chem developer and board member Prof. John Herbert on receiving the 2024-2025 Diversity Enhancement Faculty Award! You can read the official press release here:

chemistry.osu.edu
2024-2025 Diversity Enhancement Faculty Award This award recognizes a faculty member from within The Ohio State University College of Arts and Sciences whose research, teaching and/or service/outre...
0
0
14

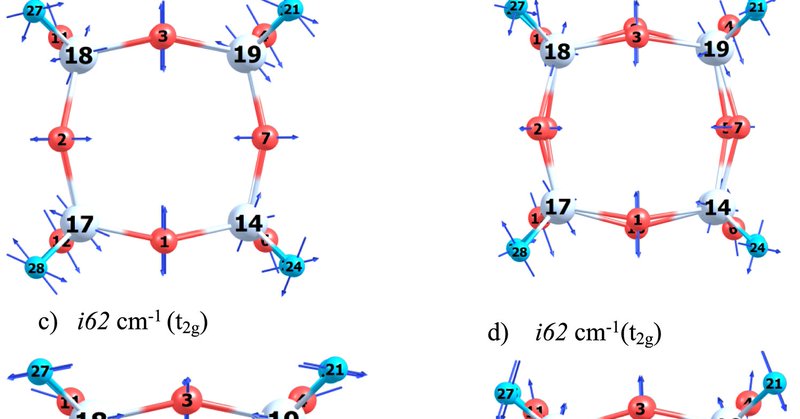
In this paper, authors use Q-Chem's geometry optimizer and DFT alongside group theory to study symmetry breaking in octahedral silsesquioxane and germanium analogues, which are used in nanohybrid functional materials. Try Q-Chem:
link.springer.com
Journal of Inorganic and Organometallic Polymers and Materials - The symmetry breaking in octahedral silsesquioxane and its germanium analogues (Si8O12H8 and Ge8O12H8) has been investigated using...
0
1
7
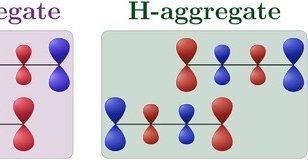
In this recent paper, authors use Q-Chem's DFT and wavefunction analysis tools to study excitonic coupling in chromophore aggregates. They present a new model for estimating the sign and magnitude of exciton coupling. Try Q-Chem:
chemistry-europe.onlinelibrary.wiley.com
Exciton coupling in organic chromophores is revisited through the lens of the transition density. The presented formalism gives insight into the strength and sign of the coupling based on the...
0
1
11
Don't miss tomorrow's webinar from Mathew Chow! He will discuss NEO-DFT and real-time NEO-TDDFT methods, along with NEO quantum mechanical/molecular mechanical approaches. Register:

0
0
5
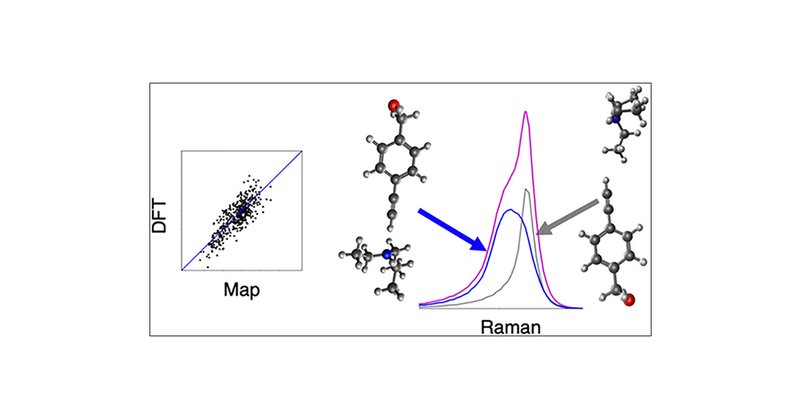
In this recent paper, researchers at Haverford College use MD alongside Q-Chem's DFT to help explain why the Raman peak for alkynes solvated in triethylamine is so unusually broad. Read the paper here: Try Q-Chem for free:
pubs.acs.org
The terminal alkyne C≡C stretch has a large Raman scattering cross section in the “silent” region for biomolecules. Experimental work taking advantage of this property provide an impetus for the...
0
1
2
Join us next week for the 2025 Wormit Award Webinar, to be presented by awardee Mathew Chow! He will discuss NEO-DFT and real-time NEO-TDDFT methods, along with NEO quantum mechanical/molecular mechanical approaches. Register here:

0
0
8
As the academic year starts back up again, don't forget about Q-Chem teaching and learning resources! We provide a variety of free resources on our website, including labs, webinars, and course materials from previous Q-chem workshops. Check it out here:

0
0
5
Researchers at UCLA synthesized a series of gold carbene-metal-amide complexes, using experiment and theory to explore the photophysics and dynamics of tautomerization. They used Q-Chem's DFT and TDDFT features. Try Q-Chem:
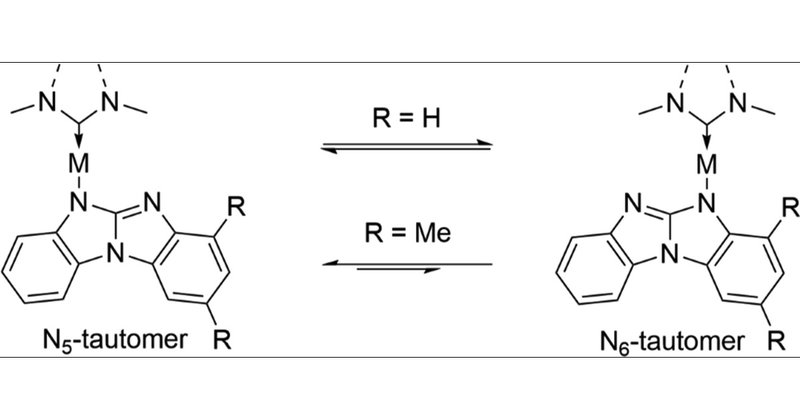
pubs.acs.org
A series of Au(I) carbene-metal-amide (cMa) complexes using N-heterocyclic carbenes and N-benzo[d]benzo[4,5]imidazo[1,2-a]imidazolyl (bim) amide ligands have been synthesized as models to investigate...
0
1
6
WebMO 25 is now available! It has a variety of new, exciting features that make its web-based interface to computational chemistry packages even better—and best of all, it now includes support for our latest release, Q-Chem 6.3! Try it out today:
0
0
6
In this recent work, researchers compare MM and QM/MM approaches to conformational sampling for the calculation of redox properties. They use Q-Chem for the QM/MM single-point energy calculations.
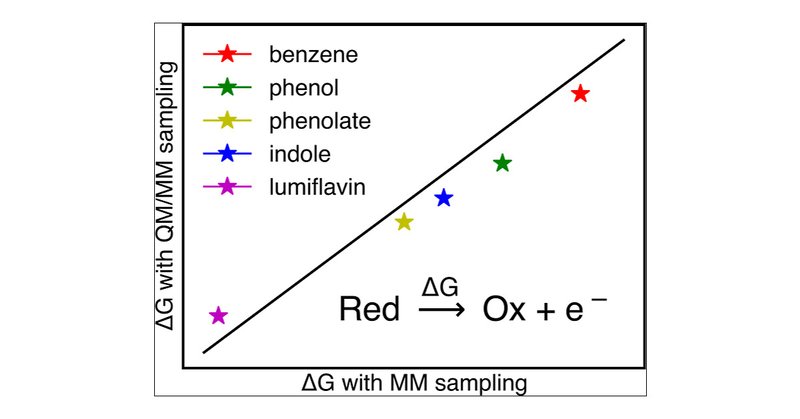
pubs.acs.org
Redox processes are an important step in many chemical and biochemical reactions. One simple approach to calculate the free energy change of a redox process is linear response approximation (LRA)....
0
4
18
Researchers use a #neuralnetwork to study how the chemical hardness of neurochemicals relates to their affinities for neuroreceptors, specifically focusing on neurotransmitters and antidepressants. They use Q-Chem for their #DFT calculations.
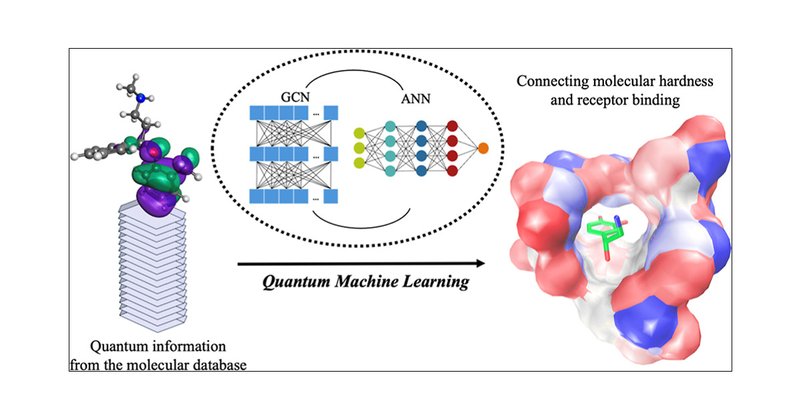
pubs.acs.org
With the advancement of artificial intelligence-embedded methodologies, their application to predict fundamental molecular properties has become increasingly prevalent. In this study, a graph...
0
0
7
Join us on July 31 at 11AM for our Nick Besley Award Webinar! The webinar will be presented by award winner Prof. Justin Talbot; he will be discussing his recent work on developing methods for studying excited state potential energy surfaces. Register:

0
0
7
Q-Chem was honored to be one of the sponsors of the 2025 MERCURY Conference at UPitt last week! We were excited to hear the multitude of ways that many of these students and professors are using Q-Chem in their ongoing research work. Thanks to those who stopped by!


0
0
5
RT @XiaoLiu161147: Truly honored and humbled that our work has been selected as a 2025 HOT @PCCP article and made free to access! Very grat….

pubs.rsc.org
Accurate prediction of barrier heights and reaction energies is of paramount importance for reaction kinetics. For computational efficiency, such calculations are typically performed with density...
0
1
0
“This workflow provides crucial insights into when Kohn-Sham DFT and even gold-standard CCSD(T) may be unreliable, particularly in the presence of strong static correlation.”. Read the paper: Try Q-Chem for free: (4/4).
pubs.rsc.org
Accurate prediction of barrier heights and reaction energies is of paramount importance for reaction kinetics. For computational efficiency, such calculations are typically performed with density...
0
0
0
“We propose our orbital stability analysis-based 3-tier classification workflow as a best-practice protocol for DFT calculations in chemical kinetics.” (3/4).
1
0
0
"By analyzing spin-symmetry breaking at both the SCF and κ-OOMP2 levels, we classified reactions based on the nature and extent of electron correlation, traced key sources of error, and addressed them to the degree possible." (2/4).
1
0
0
Congrats to Q-Chem developers and collaborators on their PCCP Hot Paper!. From author Xiao Liu: "In this work, we present a thorough investigation into the unexpectedly large RMSDs for the ωB97X-D3 functional previously reported on the RDB7 dataset (~12,000 reactions). " (🧵1/4)

1
0
6
This week, we featured papers that used various Q-Chem features, including TD-DFT with SOC, X-ray spectroscopy, CAP-EOM-EA-CCSD, and efficient gradients for localized diabatic state energies and couplings. Want to try these yourself? Request a free trial:
0
0
5
Authors investigate electron scattering in nitrous oxide, using Q-Chem's CAP-EOM-EA-CCSD and ezFCF to explain features of the experimentally-observed experimental two-dimensional electron energy loss (EELS) spectra.

0
1
4