
Vytas
@VytasGapsys
Followers
380
Following
269
Media
12
Statuses
95

We have published the prelim. speaker lineup and program over at the event page, we're incredibly excited for this lineup (including a keynote from Max Welling @wellingmax)!
1
12
23

🥳🎉 Registration for the Hünfeld #Workshop "Computer Simulation and #Theory of #Macromolecules" is now open! Don't miss your chance to join us! For more details see 🔗 https://t.co/blTupgmCot
1
24
56

Reminder: Our nonprofit, the Open Molecular Software Foundation, is hiring a research software engineer and a technical lead. Please encourage talented science software folks to apply!
0
9
19
Registration to the 2024 Workshop on Free Energy Methods in Drug Design is now open: https://t.co/r050FpycPb

ssl.eventilla.com
The 2024 edition of the Free Energy Workshop will take place in Leiden, Netherlands, 13-15th May, 2024! We have not yet opened registration, but you can already book the dates in your calendars. Keep an eye on the wiki for further information: https://t.co/OGRFzVzLH7
#alchemy2024
0
12
23
Introducing the organizing committee for #alchemy2024: Barbara Zarzycka, Bert de Groot, David Hahn, Dima Lupyan, @HighSpeedMode, @hugogdtc, @jenkescheen, @VytasGapsys, @Willemjespers
0
0
11
The 2024 edition of the Free Energy Workshop will take place in Leiden, Netherlands, 13-15th May, 2024! We have not yet opened registration, but you can already book the dates in your calendars. Keep an eye on the wiki for further information: https://t.co/OGRFzVzLH7
#alchemy2024
1
22
83
Thanks everyone involved! It was great to work together on this. What we built here is an automated workflow for free energy calculations: put ligand SMILES in, get dG back. With this in hand we probed how various docking protocols would affect dG accuracy

.@VytasGapsys and @jhmchem and colleagues develop an end-to-end relative free energy workflow based on a non-equilibrium switching approach that calculates the binding free energies starting from SMILES strings. https://t.co/fLQrN6SIKQ
2
0
17
Finally, we tested the approach prospectively and in this computational exercise it looked promising (~3 kcal/mol improvement on the previous best binder). Next is to try it in a real drug discovery project.
0
0
1
We were also able to control the way in which to explore the chemical libraries. By changing ligand selection strategy we could a) quickly find a deep local minimum of strong binders or b) obtain a good overall description of the chemical space
1
0
1
First, we needed to calibrate the active learning protocol: in our hands, predictions based only on simple ligand descriptors outperformed any other fancier type of representation.
1
0
3
Our latest work on alchemical free energies. This time we used them as an oracle in an active learning loop to navigate chemical space in search for high affinity binders. Thanks to Yuriy, @gtresadern, David and Bert! https://t.co/J3qnReB28P Several main findings below

pubs.acs.org
Drug discovery can be thought of as a search for a needle in a haystack: searching through a large chemical space for the most active compounds. Computational techniques can narrow the search space...
3
6
64

<- Computational Chemist with background in Quantum Chemistry, protein Molecular Dynamics and industrial experience in Material Science searching for a new position. Ideally in the Karlsruhe/Heidelberg area but not limited to it. Find my LinkedIn profile:
de.linkedin.com
The research field of computational biophysical chemistry provides an interdisciplinary… · Berufserfahrung: Merck Healthcare · Ausbildung: Georg-August-Universität Göttingen · Ort: Bruchsal · 456...
1
9
26

Pre-Exascale Computing of Protein–Ligand Binding #FreeEnergies with Open Source Software for #DrugDesign #cheminformatics
https://t.co/YZqgwqoyyq
@davidlmobley @gtresadern
#current_issue #JCIM #Letters
0
15
70
@JCIM_JCTC Thanks for playing. The answer is: 1. The use of maps is not allowed. 2. Clipart is not allowed, unless it is from the Microsoft Suite. I understand that this is probably licensing policy, but feels weird that showing contours of continents is forbidden
1
0
4
Here is something unexpected: just discovered several hidden rules for the @JCIM_JCTC table of contents figure. Can you guess what two rules are violated by this TOC image?
4
0
7
As a result, we also made free energy predictions for the systems in @Ch_Schindler's dataset with the open source force fields, including @openforcefield
0
0
3
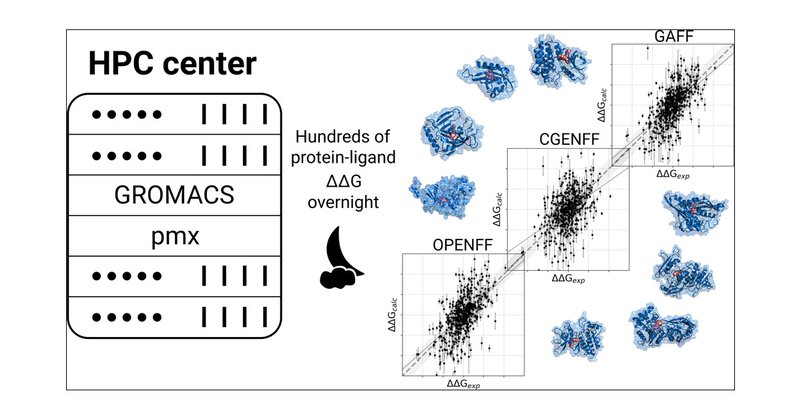
It was great to see that the alchemical protocols parallelize really efficiently: we could make use of ~500 nodes of the Max Planck new supercomputer 'Raven' for 3 days (more than 3 million core hours).
2
0
6
Our latest study on ligand alchemy now in @JCIM_ACS with David @gtresadern @davidlmobley Markus and Bert: https://t.co/DEWZZRJLaF The question we asked here: how far can we push the limits of scaling such calculations relying on the open source software and force fields?
pubs.acs.org
Nowadays, drug design projects benefit from highly accurate protein–ligand binding free energy predictions based on molecular dynamics simulations. While such calculations have been computationally...
2
17
85